| Size | Price | Stock | Qty |
|---|---|---|---|
| 1mg |
|
||
| 5mg |
|
||
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| Other Sizes |
|
Purity: ≥98%
APX001A (Manogepix, E1210) is a first-in-class, broad-spectrum and orally bioactive inhibitor of the fungal protein Gwt1 (GPI-anchored wall transfer protein 1) in the glycosylphosphatidylinositol (GPI) biosynthesis pathway. Gwt1 protein is an enzyme required for acylation of inositol during glycosylphosphatidylinositol anchor biosynthesis. Manogepix (APX001A) is the active metabolite of fosmanogepix (APX001), which is a drug candidate currently in clinical trials for the treatment of invasive fungal infections. APX001A is active against the major fungal pathogens, i.e., Candida (except Candida krusei), Aspergillus, and hard-to-treat molds, including Fusarium and Scedosporium.
| Targets |
Antifungal
|
|---|---|
| ln Vitro |
Manogepix has no inhibitory effect against human Pig-Wp even at concentrations as high as 100 μM, but it does reduce the inositol acylation activity of C. albicans Gwt1p and A. fumigatus Gwt1p with IC50s of 0.3 to 0.6 μM. The expression of GPI-anchored protein ALS1 on the surfaces of Candida albicans cells treated with Manogepix is examined and shown to be much lower than that on untreated cells, confirming the inhibition of fungal glycosylphosphatidylinositol (GPI) biosynthesis. At concentrations above its minimum inhibitory concentration (MIC), manogepix inhibits the production of germ tubes, adhesion to polystyrene surfaces, and biofilm formation of Candida albicans [1].
|
| ln Vivo |
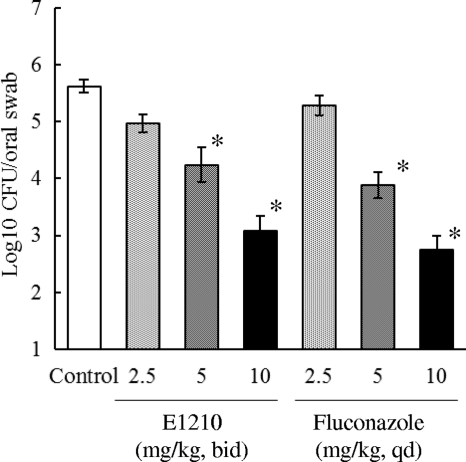
The quantity of viable C is decreased by manogepix treatment (oral administration; twice daily; for three days; specific-pathogen-free female ICR mice; 2.5 mg/kg, 5 mg/kg, and 10 mg/kg). oral cavity albicans cells in a dose-dependent way[2].
|
| Enzyme Assay |
In vitro susceptibility testing. [2]
The MICs of E1210 and the reference compounds were determined using the broth microdilution method detailed by the Clinical and Laboratory Standards Institute (CLSI) in documents M27-A3 and M38-A2. RPMI 1640 medium buffered to pH 7.0 with 0.165 M 3-(N-morpholino)-propanesulfonic acid (MOPS) was used. The results were expressed as the median MIC of each compound derived from three independent experiments. Continued research toward the development of new antifungals that act via inhibition of glycosylphosphatidylinositol (GPI) biosynthesis led to the design of E1210. In this study, we assessed the selectivity of the inhibitory activity of E1210 against Candida albicans GWT1 (Orf19.6884) protein, Aspergillus fumigatus GWT1 (AFUA_1G14870) protein, and human PIG-W protein, which can catalyze the inositol acylation of GPI early in the GPI biosynthesis pathway, and then we assessed the effects of E1210 on key C. albicans virulence factors. E1210 inhibited the inositol acylation activity of C. albicans Gwt1p and A. fumigatus Gwt1p with 50% inhibitory concentrations (IC(50)s) of 0.3 to 0.6 μM but had no inhibitory activity against human Pig-Wp even at concentrations as high as 100 μM. To confirm the inhibition of fungal GPI biosynthesis, expression of ALS1 protein, a GPI-anchored protein, on the surfaces of C. albicans cells treated with E1210 was studied and shown to be significantly lower than that on untreated cells. However, the ALS1 protein levels in the crude extract and the RHO1 protein levels on the cell surface were found to be almost the same. Furthermore, E1210 inhibited germ tube formation, adherence to polystyrene surfaces, and biofilm formation of C. albicans at concentrations above its MIC. These results suggested that E1210 selectively inhibited inositol acylation of fungus-specific GPI which would be catalyzed by Gwt1p, leading to the inhibition of GPI-anchored protein maturation, and also that E1210 suppressed the expression of some important virulence factors of C. albicans, through its GPI biosynthesis inhibition.[1] |
| Cell Assay |
The Als1p levels in crude extracts of C. albicans cells were also determined. The cells were incubated for 1 h at 35°C in the presence of E1210. After incubation, the cultures were centrifuged at 1,000 × g for 10 min at 4°C and the pellets were suspended in 50 mM potassium phosphate buffer (pH 7.4) containing a protease inhibitor cocktail for fungi (Sigma-Aldrich). The cell suspensions were mixed with an equal weight of glass beads and were homogenized with the cell disruptor. The unbroken cells and glass beads were then removed by centrifugation. The supernatants were obtained as crude extracts. The amount of Als1 protein in the crude extracts was also measured by an ELISA. Anti-Als1 antibody was used as the capture antibody, and anti-C. albicans, HRP-conjugated rabbit antibody (ViroStat Inc., Portland, ME) was used as the secondary antibody. An ELISA of each concentration was conducted in duplicate, and the final values were expressed as the means of three determinations. Protein concentration was estimated in order to standardize the amount of Als1p.[1]
|
| Animal Protocol |
Animal/Disease Models: Specific-pathogen-free female ICR mice (5 weeks; ~25 g) with C. albicans[2]
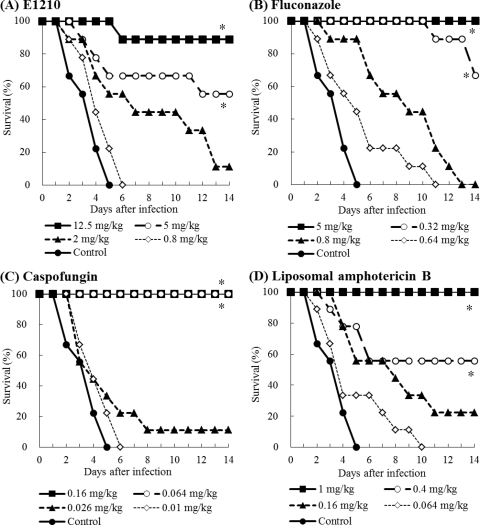
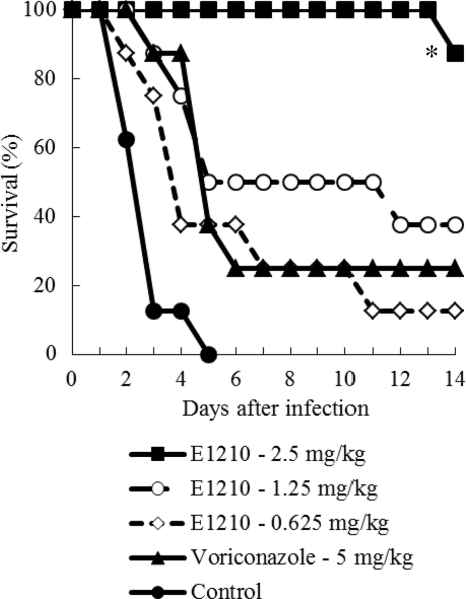
Doses: 2.5 mg/kg, 5 mg/kg and 10 mg /kg Route of Administration: Oral administration; twice (two times) daily; for 3 days Experimental Results: decreased the number of viable C. albicans cells in the oral cavity in a dose-dependent manner. Oropharyngeal candidiasis model. [2] C. albicans was used to infect mice that were immunosuppressed with cortisone, and the number of C. albicans cells in the oral cavity of each mouse was measured following drug treatment. ICR mice were immunosuppressed using 4 mg of subcutaneously administered cortisone acetate given 1 day before and 3 days after infection. The mice were also given 1 mg/ml tetracycline hydrochloride via their drinking water, starting on the day of cortisone administration and continuing throughout the experiment, in order to prevent bacterial infection. C. albicans IFM49971 was grown on Sabouraud dextrose agar (SDA) at 35°C for 2 days. The cells were suspended in sterile normal saline. The cells were counted with a hemocytometer and adjusted to the required density with sterile normal saline. The mice were then anesthetized with chlorpromazine hydrochloride (0.5 mg/mouse given subcutaneously). By use of a micropipette, aliquots (10 μl) of C. albicans IFM49971 suspension were inoculated into the oral cavities of the anesthetized mice. Then the challenge dose of 4 × 105 CFU of C. albicans (CFU)/mouse was given. This was followed with either E1210 orally administered twice daily (BID) or fluconazole orally administered once daily (QD) for three consecutive days starting 3 days after infection. The control group was given the equivalent volume of 5% glucose BID. The mice were anesthetized with chlorpromazine hydrochloride (0.5 mg/mouse subcutaneously) the day after the final dose of the study drug. Efficacy was assessed by determination of the number of C. albicans cells in the oral cavity of each mouse after study drug treatment. The oral cavity (that is, the cheek, tongue, and soft palate) was thoroughly swabbed using a fine-tipped cotton swab. After swabbing, the cotton end was placed into a test tube containing 1 ml sterile normal saline. The cells recovered were suspended in sterile normal saline by mixing them on a vortex mixer before being cultured, after serial 10-fold dilutions, on SDA plates supplemented with ampicillin (0.1 mg/ml). The SDA plates were incubated at 35°C overnight, and then the viable cells were counted as the number of CFU. The cell number was expressed in units of log10 CFU/swab. The lowest detectable number of cells in the oral cavity was 10 CFU (1 log10 CFU). The viable cell counts were performed in duplicate.[2] Disseminated candidiasis model. [2] ICR mice were immunosuppressed utilizing 5-fluorouracil (5-FU) at 200 mg/kg of body weight subcutaneously administered 6 days prior to infection. These mice were also administered 0.1 mg/ml ciprofloxacin orally via their drinking water, from 2 to 3 days prior to infection to 5 to 7 days after infection, in order to prevent endogenous bacterial infections. C. albicans IFM49971, C. albicans IFM49738, and C. tropicalis E83037 were each cultured on an SDA plate at 35°C for 2 days. The cells from the surface of the agar plate were suspended in sterile normal saline, and the cells were counted with a hemocytometer. The final inoculum was adjusted to the required density using sterile normal saline. Infection was induced in the neutropenic mice by the intravenous administration of 0.2 ml of a C. albicans cell suspension (0.8 to 1.4 × 104 CFU/mouse or 5.3 × 104 CFU/mouse for IFM49971) or of a C. tropicalis cell suspension (3.0 × 105 CFU/mouse) injected into the lateral tail vein. Antifungal therapy was initiated 1 h or 24 h after infection and was continued for three consecutive days (days 0 to 2 or 1 to 3). E1210 or voriconazole was each orally administered two or three times daily, fluconazole was orally administered once daily, and caspofungin or liposomal amphotericin B was intravenously administered once daily. The control group received an equivalent volume of vehicle (5% glucose, 10 ml/kg) orally two or three times daily. In our preliminary studies, the survival curve of control mice receiving vehicle orally was similar to that of mice receiving vehicle intravenously. Therefore, we did not set up the control group to receive vehicle intravenously. The survival rate and survival period were determined over 14 days.[2] View More
Pulmonary aspergillosis model. [2] Disseminated fusariosis model. [2] DBA/2N mice were immunosuppressed with 200 mg/kg of subcutaneously administered 5-FU, 6 days prior to infection. The mice were also administered 0.1 mg/ml ciprofloxacin orally in their drinking water, from 3 days prior to infection until 7 days after infection, to prevent bacterial infections. F. solani IFM50956 was cultured on a PDA plate at 30°C for 7 days. The cells from the surface of the agar plate were suspended in sterile normal saline containing 0.05% Tween 80, and the cells were counted using a hemocytometer. The final inoculum was adjusted to the required density using sterile normal saline containing 0.05% Tween 80. Infection was induced in the neutropenic mice by the intravenous inoculation of a 0.2-ml F. solani cell suspension (5.0 × 103 cells/mouse) into the lateral tail vein. Antifungal therapy was initiated 1 h after infection and was continued for five consecutive days (days 0 to 4). E1210 was orally administered three times a day (TID). The control group received an equivalent volume of 5% glucose orally TID. The survival rate and survival period were determined over 14 days.[2] Pharmacokinetic study. [2] E1210 was intravenously or orally administered to male ICR mice. After administration of E1210, blood samples were drawn from the vena cava of each mouse at designated time points (0.08, 0.25, 0.5, 1, 2, 4, 6, 8 h). Plasma samples were obtained by centrifuging blood. After deproteinization with methanol, the extracted sample was analyzed by liquid chromatography-tandem mass spectrometry (LC/MS/MS). The concentrations of E1210 in plasma were determined by an internal standard method using MassLynx. The pharmacokinetic parameters of E1210 were calculated by model independent analysis.[2] Toxicology study. [2] E1210 was administered orally by gavage once a day for 7 days to male and female Sprague-Dawley rats (3 animals/group/gender) at doses of 100, 300, or 1,000 mg/kg. A control group received an equivalent volume (10 ml/kg) of vehicle (0.4 mol/liter hydrochloric acid). All rats found dead or moribund were necropsied as soon as they were discovered, and all surviving animals were necropsied after 7 days of administration. The following were evaluated: mortality, clinical signs, body weight, food consumption, hematology, blood chemistry, toxicokinetics, hepatic drug-metabolizing enzymes, and macroscopic and microscopic pathologies.[2] |
| References |
|
| Additional Infomation |
E1210 is a first-in-class, broad-spectrum antifungal with a novel mechanism of action-inhibition of fungal glycosylphosphatidylinositol biosynthesis. In this study, the efficacies of E1210 and reference antifungals were evaluated in murine models of oropharyngeal and disseminated candidiasis, pulmonary aspergillosis, and disseminated fusariosis. Oral E1210 demonstrated dose-dependent efficacy in infections caused by Candida species, Aspergillus spp., and Fusarium solani. In the treatment of oropharyngeal candidiasis, E1210 and fluconazole each caused a significantly greater reduction in the number of oral CFU than the control treatment (P < 0.05). In the disseminated candidiasis model, mice treated with E1210, fluconazole, caspofungin, or liposomal amphotericin B showed significantly higher survival rates than the control mice (P < 0.05). E1210 was also highly effective in treating disseminated candidiasis caused by azole-resistant Candida albicans or Candida tropicalis. A 24-h delay in treatment onset minimally affected the efficacy outcome of E1210 in the treatment of disseminated candidiasis. In the Aspergillus flavus pulmonary aspergillosis model, mice treated with E1210, voriconazole, or caspofungin showed significantly higher survival rates than the control mice (P < 0.05). E1210 was also effective in the treatment of Aspergillus fumigatus pulmonary aspergillosis. In contrast to many antifungals, E1210 was also effective against disseminated fusariosis caused by F. solani. In conclusion, E1210 demonstrated consistent efficacy in murine models of oropharyngeal and disseminated candidiasis, pulmonary aspergillosis, and disseminated fusariosis. These data suggest that further studies to determine E1210's potential for the treatment of disseminated fungal infections are indicated.[2]
|
| Molecular Formula |
C21H18N4O2
|
|---|---|
| Molecular Weight |
358.4
|
| Exact Mass |
358.142
|
| Elemental Analysis |
C, 70.38; H, 5.06; N, 15.63; O, 8.93
|
| CAS # |
936339-60-5
|
| Related CAS # |
Fosmanogepix;2091769-17-2
|
| PubChem CID |
16719049
|
| Appearance |
White to light yellow solid powder
|
| Density |
1.3±0.1 g/cm3
|
| Boiling Point |
569.4±45.0 °C at 760 mmHg
|
| Flash Point |
298.1±28.7 °C
|
| Vapour Pressure |
0.0±1.6 mmHg at 25°C
|
| Index of Refraction |
1.640
|
| LogP |
3.75
|
| Hydrogen Bond Donor Count |
1
|
| Hydrogen Bond Acceptor Count |
6
|
| Rotatable Bond Count |
6
|
| Heavy Atom Count |
27
|
| Complexity |
444
|
| Defined Atom Stereocenter Count |
0
|
| InChi Key |
WSEKTEUGRLFBSE-UHFFFAOYSA-N
|
| InChi Code |
InChI=1S/C21H18N4O2/c22-21-18(4-3-11-24-21)19-13-17(25-27-19)12-15-6-8-16(9-7-15)14-26-20-5-1-2-10-23-20/h1-11,13H,12,14H2,(H2,22,24)
|
| Chemical Name |
(3-(3-(4-(((Pyridin-2-yl)oxy)methyl)benzyl)isoxazol-5-yl)pyridin-2-)amine
|
| Synonyms |
E1210; APX001A; E-1210; APX 001 A; APX001A; E1210; Manogepix; E-1210; 3-(3-(4-((Pyridin-2-yloxy)methyl)benzyl)isoxazol-5-yl)pyridin-2-amine; (3-(3-(4-(((Pyridin-2-yl)oxy)methyl)benzyl)isoxazol-5-yl)pyridin-2-)amine; 7B1P18ID9L; E 1210; APX-001-A; APX001-A; Manogepix
|
| HS Tariff Code |
2934.99.9001
|
| Storage |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| Solubility (In Vitro) |
DMSO : ~100 mg/mL (~279.03 mM)
|
|---|---|
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.08 mg/mL (5.80 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution.
For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.8 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2.08 mg/mL (5.80 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.8 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. View More
Solubility in Formulation 3: ≥ 2.08 mg/mL (5.80 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.7902 mL | 13.9509 mL | 27.9018 mL | |
| 5 mM | 0.5580 mL | 2.7902 mL | 5.5804 mL | |
| 10 mM | 0.2790 mL | 1.3951 mL | 2.7902 mL |
*Note: Please select an appropriate solvent for the preparation of stock solution based on your experiment needs. For most products, DMSO can be used for preparing stock solutions (e.g. 5 mM, 10 mM, or 20 mM concentration); some products with high aqueous solubility may be dissolved in water directly. Solubility information is available at the above Solubility Data section. Once the stock solution is prepared, aliquot it to routine usage volumes and store at -20°C or -80°C. Avoid repeated freeze and thaw cycles.
Calculation results
Working concentration: mg/mL;
Method for preparing DMSO stock solution: mg drug pre-dissolved in μL DMSO (stock solution concentration mg/mL). Please contact us first if the concentration exceeds the DMSO solubility of the batch of drug.
Method for preparing in vivo formulation::Take μL DMSO stock solution, next add μL PEG300, mix and clarify, next addμL Tween 80, mix and clarify, next add μL ddH2O,mix and clarify.
(1) Please be sure that the solution is clear before the addition of next solvent. Dissolution methods like vortex, ultrasound or warming and heat may be used to aid dissolving.
(2) Be sure to add the solvent(s) in order.
| NCT Number | Recruitment | interventions | Conditions | Sponsor/Collaborators | Start Date | Phases |
| NCT05491733 | Completed | Drug: APX001 Drug: APX001A |
Invasive Fungal Infections | Basilea Pharmaceutica | 2021-03-02 | Phase 1 |
| NCT04166669 | Completed | Drug: APX001 Drug: Itraconazole Drug: Rifampin |
Fungal Infection | Basilea Pharmaceutica | 2019-11-12 | Phase 1 |
| NCT04240886 | Terminated | Drug: fosmanogepix | Invasive Fungal Infections | Basilea Pharmaceutica | 2020-01-04 | Phase 2 |
| NCT05582187 | Recruiting | Drug: Fosmanogepix | Hepatic Impairment | Basilea Pharmaceutica | 2022-10-31 | Phase 1 |
|
|
|
Products are for research use only; We do not sell to patients
Copyright 2020 InvivoChem LLC | All Rights Reserved






















































 COA
COA