| Size | Price | Stock | Qty |
|---|---|---|---|
| 50mg |
|
||
| 100mg |
|
||
| 500mg |
|
||
| 1g |
|
||
| 2g |
|
||
| 5g |
|
||
| Other Sizes |
|
































Purity: ≥98%
Atorvastatin (CI-981; CI981; Tozalip; Torvast; Cardyl; liptonorm) is an approved blockbuster drug of the statin class used as an LDL
cholesterol-lowering/hypolipidemic medication. It act as a potent and
selective inhibitor of HMG-CoA reductase.Atorvastatin isthe best-selling drug in the history of pharmaceuticals with a total of around US$130 billion for Pfizer during its 14 years on the market, making it the world's bestselling drug of all time. It acts by blocking the production of cholesterol. Atorvastatin is used primarily for lowering blood cholesterol and for prevention of events associated with cardiovascular disease.
| Targets |
HMG-CoA reductase
|
|---|---|
| ln Vitro |
By downregulating the expression of GRP78, caspase-12, and CHOP in cardiomyocytes during myocardial infarction, atorvastatin treatment lowers cardiomyocyte apoptosis. Additionally, it stimulates the endoplasmic reticulum (ER) in response to heart failure and angiotensin II (Ang II) stimulation. ) tension. 4].
|
| ln Vivo |
Treatment with atorvastatin (20–30 mg/kg; oral gavage; once daily; for 28 days; ApoE−/− mice) markedly decreased the amount of apoptotic cells, endoplasmic reticulum (ER) stress signaling proteins, and Caspase12 and Caspase12 activation. Bax in ApoE-/- mice triggered by Ang II. Following atorvastatin treatment, pro-inflammatory cytokines such IL-6, IL-8, and IL-1β were markedly suppressed [5].
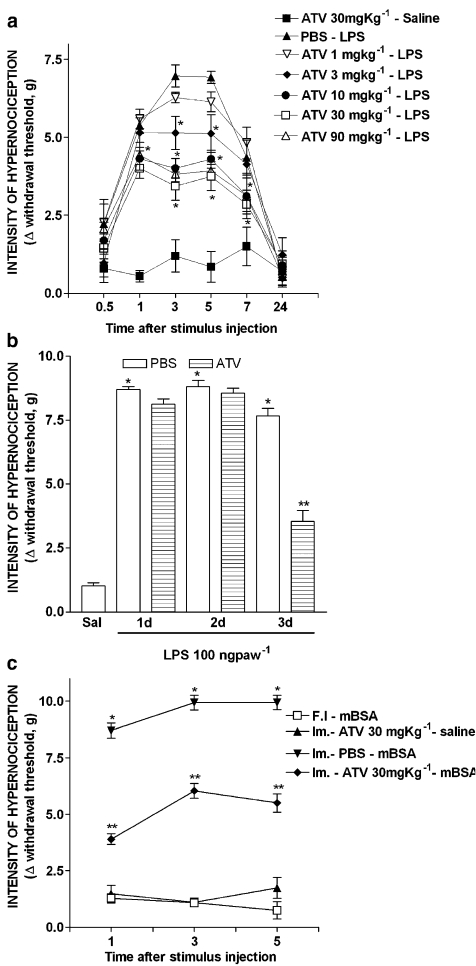
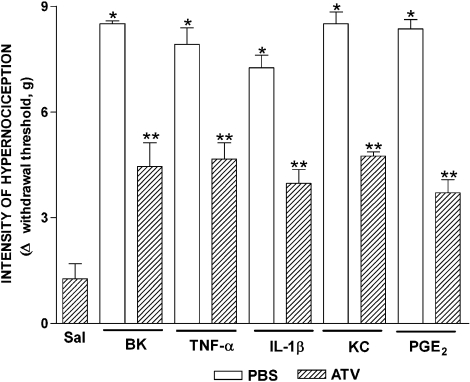
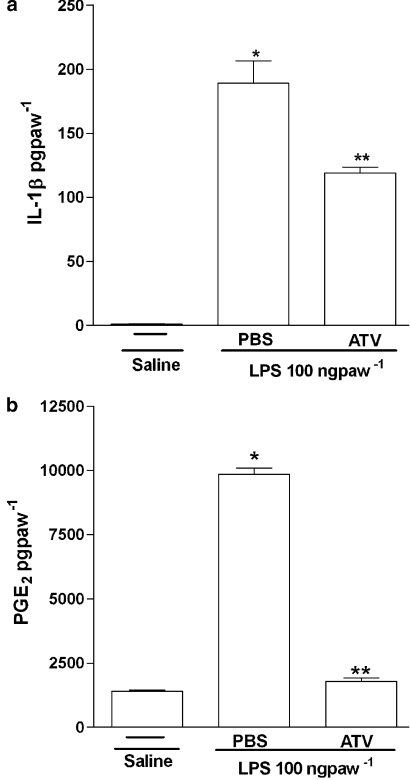
The effects of orally administered atorvastatin on inflammatory mechanical hypernociception in mouse paws were evaluated with an electronic pressure-meter. Cytokines and PGE(2) were measured by ELISA and RIA. Key results: Treatment with atorvastatin for 3 days dose-dependently reduced hypernociception induced by lipopolysaccharide (LPS) or that following antigen challenge in sensitized animals. Atorvastatin pre-treatment reduced hypernociception induced by bradykinin and cytokines (TNF-alpha, IL-1beta and KC), and the release of IL-1beta and PGE(2) in paw skin, induced by lipopolysaccharide. The antinociceptive effect of atorvastatin on LPS-induced hypernociception was prevented by mevalonate co-treatment without affecting serum cholesterol levels. Hypernociception induced by PGE(2) was inhibited by atorvastatin, suggesting intracellular antinociceptive mechanisms for atorvastatin. The antinociceptive effect of atorvastatin upon LPS- or PGE(2)-induced hypernociception was prevented by non-selective inhibitors of nitric oxide synthase (NOS) but not by selective inhibition of inducible NOS or in mice lacking this enzyme.[1] |
| Enzyme Assay |
The HMG-CoA reductase assay kit with the catalytic domain of the human enzyme (recombinant GST fusion protein expressed in E. coli) was used, under conditions recommended by the manufacturer, to identify the most effective fraction of plant extract. The concentration of the purified human enzyme stock solution was 0.52–0.85 mg protein/mL. Reference statin drug pravastatin was used as positive control. To characterize HMG-CoA reductase inhibition under defined assay conditions, reactions containing 4 μL of NADPH (to obtain a final concentration of 400 μM) and 12 μL of HMG-CoA substrate (to obtain a final concentration of 400 μM) in a final volume of 0.2 mL of 100 mM potassium phosphate buffer, pH 7.4 (containing 120 mM KCl, 1 mM EDTA, and 5 mM DTT), were initiated (time 0) by the addition of 2 μL of the catalytic domain of human recombinant HMG-CoA reductase and incubated in Eppendorf BioSpectrometer (equipped with thermostatically controlled cell holder) at 37°C in the presence or absence (control) of 1 μL aliquots of drugs dissolved in DMSO. The rates of NADPH consumed were monitored every 20 sec for up to 15 min by scanning spectrophotometrically [7].
|
| Cell Assay |
Cell proliferation assays were performed essentially as described previously. Briefly, SV-SMC from 5 different patients were seeded into 24-well cell culture plates at a density of 1 × 104 cells per well in full growth medium. Cells were incubated overnight and then quiesced in serum free medium for 3 days before transfer to full growth medium (10% FCS) containing 5 different statins at a range of concentrations. All statins were tested on cells from each individual patient. Medium and drugs were replaced after 2 days, and viable cell numbers were determined in triplicate wells after 4 days using Trypan Blue and a hemocytometer. The increase in cell number was calculated by subtracting the starting cell number (day 0) from the final cell number (day 4). Data were then normalized to control values (no statin) to correct for differences in proliferation rates between cells from different patients [2].
|
| Animal Protocol |
Animal/Disease Models: 40 8weeks old ApoE−/− mice, angiotensin II (Ang II) induced [5]
Doses: 20 mg/kg, 30 mg/kg Route of Administration: po (oral gavage); one time/day; continuous 28-day Experimental Results: Endoplasmic reticulum stress signaling proteins, the number of apoptotic cells, and the activation of Caspase12 and Bax were Dramatically diminished in Ang II-induced ApoE−/− mice. Pro-inflammatory cytokines such as IL-6, IL-8, and IL-1β were Dramatically inhibited. Effect of atorvastatin on hypernociception induced by LPS or antigen challenge [1] To investigate the effect of atorvastatin on lipopolysaccharide (LPS)-induced inflammatory hypernociception, mice were pretreated orally with either atorvastatin, at doses of 1, 3, 10, 30 and 90 mg kg−1 or vehicle (PBS) once a day for 3 consecutive days. At 2 h after the last dose of atorvastatin, mice received an i.pl. injection of LPS (100 ng paw−1) or saline (vehicle for LPS). The animals were also treated with atorvastatin (30 mg kg−1) for 1 or 2 days before LPS challenge. The hypernociceptive responses were assessed 0.5, 1, 3, 5, 7 and 24 h after LPS or saline i.pl. injections. In addition, we investigated the effect of atorvastatin on the immune inflammatory hypernociception in mice sensitized to mBSA and challenged with antigen. The animals were pretreated orally with atorvastatin (30 mg kg−1) or PBS once a day for 3 consecutive days. At 2 h after the last dose of atorvastatin, mice received an i.pl. injection of mBSA (90 μg paw−1) or saline. In the control group, mBSA was injected into the paws of the false immunized mice (see above). Mice were fasted for 8 h receiving atorvastatin or PBS. The hypernociceptive responses were assessed 1, 3 and 5 h after challenge with antigen. Effect of atorvastatin on hypernociception induced by bradykinin, cytokines or PGE2 [1] In this set of experiments, the effect of atorvastatin was investigated on mechanical hypernociception induced by bradykinin (BK) (500 ng paw−1), TNF-α (50 pg paw−1), IL-1β (1 ng paw−1), keratinocyte-derived chemokine (KC/CXCL) (20 ng paw−1) and PGE2 (100 ng paw−1). The animals were pretreated for 3 days with atorvastatin (30 mg kg−1, peritoneally (p.o.)) or PBS, as described above. Hypernociception was assessed 3 h after injection of the inflammatory stimulus (or saline) in the paw. Effect of atorvastatin on IL-1β and PGE2 production induced by LPS [1] To investigate whether the antinociceptive effect of atorvastatin depended on the inhibition of IL-1β and PGE2 production induced by LPS, the levels of these mediators were measured in the paw skin of mice pretreated for 3 days with atorvastatin (30 mg kg−1 p.o.) or PBS, as described above. The levels of these mediators in paw skin were determined 3 h after injection of LPS or saline into the paw. Influence of NOS inhibitors on the antinociceptive effect of atorvastatin [1] To assess the contribution of NO to the antinociceptive effect of atorvastatin, animals were pretreated with the statin, as described above. One hour before the injection of LPS or PGE2 into the paw, mice received an NOS inhibitor, either l-arginine analog N-nitro-l-arginine methyl ester (l-NAME) (90 mg kg−1, i.p.), L-NMMA (90 mg kg−1, i.p.) or 1400W (1.5 mg kg−1, i.v.). In a different series of experiments, using the mice lacking iNOS (iNOS −/−) and the relevant WT mice, we assessed the effect of atorvastatin (given as described) on LPS-induced hypernociception. In both sets of experiments, hypernociception was assessed 3 h after injection of LPS- or PGE2-i.pl. Role of products of HMG-CoA reductase on the antinociceptive effect of atorvastatin [1] To investigate whether the antinociceptive effect of atorvastatin reflected decreased levels of the products of HMG-CoA, two types of experiments were performed. In one, the total serum cholesterol concentration was determined in mice treated with atorvastatin at a dose of 30 mg kg−1 day−1, or PBS, for 3 days and then injected i.pl. with LPS or saline. In the other, the HMG-CoA reductase product, mevalonate, was given (10–90 mg kg−1) at the same times as atorvastatin. Hypernociception and cholesterol levels were determined 3 h after i.pl. LPS or saline. |
| ADME/Pharmacokinetics |
Absorption, Distribution and Excretion
Atorvastatin presents a dose-dependent and non-linear pharmacokinetic profile. It is very rapidly absorbed after oral administration. After the administration of a dose of 40 mg, its peak plasma concentration of 28 ng/ml is reached 1-2 hours after initial administration with an AUC of about 200 ng∙h/ml. Atorvastatin undergoes extensive first-pass metabolism in the wall of the gut and the liver, resulting in an absolute oral bioavailability of 14%. Plasma atorvastatin concentrations are lower (approximately 30% for Cmax and AUC) following evening drug administration compared with morning. However, LDL-C reduction is the same regardless of the time of day of drug administration. Administration of atorvastatin with food results in prolonged Tmax and a reduction in Cmax and AUC. Breast Cancer Resistance Protein (BCRP) is a membrane-bound protein that plays an important role in the absorption of atorvastatin. Evidence from pharmacogenetic studies of c.421C>A single nucleotide polymorphisms (SNPs) in the gene for BCRP has demonstrated that individuals with the 421AA genotype have reduced functional activity and 1.72-fold higher AUC for atorvastatin compared to study individuals with the control 421CC genotype. This has important implications for the variation in response to the drug in terms of efficacy and toxicity, particularly as the BCRP c.421C>A polymorphism occurs more frequently in Asian populations than in Caucasians. Other statin drugs impacted by this polymorphism include [fluvastatin], [simvastatin], and [rosuvastatin]. Genetic differences in the OATP1B1 (organic-anion-transporting polypeptide 1B1) hepatic transporter encoded by the SCLCO1B1 gene (Solute Carrier Organic Anion Transporter family member 1B1) have been shown to impact atorvastatin pharmacokinetics. Evidence from pharmacogenetic studies of the c.521T>C single nucleotide polymorphism (SNP) in the gene encoding OATP1B1 (SLCO1B1) demonstrated that atorvastatin AUC was increased 2.45-fold for individuals homozygous for 521CC compared to homozygous 521TT individuals. Other statin drugs impacted by this polymorphism include [simvastatin], [pitavastatin], [rosuvastatin], and [pravastatin]. Atorvastatin and its metabolites are mainly eliminated in the bile without enterohepatic recirculation. The renal elimination of atorvastatin is very minimal and represents less than 1% of the eliminated dose. The reported volume of distribution of atorvastatin is of 380 L. The registered total plasma clearance of atorvastatin is of 625 ml/min. /MILK/ In a separate experiment, a single dose of 10 mg/kg atorvastatin administered to female Wistar rats on gestation day 19 or lactation day 13 provided evidence of placental transfer and excretion into the milk. Lipitor and its metabolites are eliminated primarily in bile following hepatic and/or extra-hepatic metabolism; however, the drug does not appear to undergo enterohepatic recirculation. ... Less than 2% of a dose of Lipitor is recovered in urine following oral administration. /MILK/ It is not known whether atorvastatin is excreted in human milk, but a small amount of another drug in this class does pass into breast milk. Nursing rat pups had plasma and liver drug levels of 50% and 40%, respectively, of that in their mother's milk. Mean volume of distribution of Lipitor is approximately 381 liters. Lipitor is >/= 98% bound to plasma proteins. A blood/plasma ratio of approximately 0.25 indicates poor drug penetration into red blood cells. For more Absorption, Distribution and Excretion (Complete) data for ATORVASTATIN (8 total), please visit the HSDB record page. Metabolism / Metabolites Atorvastatin is highly metabolized to ortho- and parahydroxylated derivatives and various beta-oxidation products, primarily by Cytochrome P450 3A4 in the intestine and liver. Atorvastatin's metabolites undergo further lactonization via the formation of acyl glucuronide intermediates by the enzymes UGT1A1 and UGT1A3. These lactones can be hydrolyzed back to their corresponding acid forms and exist in equilibirum. _In vitro_ inhibition of HMG-CoA reductase by ortho- and parahydroxylated metabolites is equivalent to that of atorvastatin. Approximately 70% of circulating inhibitory activity for HMG-CoA reductase is attributed to active metabolites. Lipitor is extensively metabolized to ortho- and parahydroxylated derivatives and various beta-oxidation products. In vitro inhibition of 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase by ortho- and parahydroxylated metabolites is equivalent to that of Lipitor. Approximately 70% of circulating inhibitory activity for HMG-CoA reductase is attributed to active metabolites. In vitro studies suggest the importance of Lipitor metabolism by cytochrome P450 3A4, consistent with increased plasma concentrations of Lipitor in humans following co-administration with erythromycin, a known inhibitor of this isozyme. In animals, the ortho-hydroxy metabolite undergoes further glucuronidation. The active forms of all marketed hydroxymethylglutaryl (HMG)-CoA reductase inhibitors share a common dihydroxy heptanoic or heptenoic acid side chain. In this study, we present evidence for the formation of acyl glucuronide conjugates of the hydroxy acid forms of simvastatin (SVA), atorvastatin (AVA), and cerivastatin (CVA) in rat, dog, and human liver preparations in vitro and for the excretion of the acyl glucuronide of SVA in dog bile and urine. Upon incubation of each statin (SVA, CVA or AVA) with liver microsomal preparations supplemented with UDP-glucuronic acid, two major products were detected. Based on analysis by high-pressure liquid chromatography, UV spectroscopy, and/or liquid chromatography (LC)-mass spectrometry analysis, these metabolites were identified as a glucuronide conjugate of the hydroxy acid form of the statin and the corresponding delta-lactone. By means of an LC-NMR technique, the glucuronide structure was established to be a 1-O-acyl-beta-D-glucuronide conjugate of the statin acid. The formation of statin glucuronide and statin lactone in human liver microsomes exhibited modest intersubject variability (3- to 6-fold; n = 10). Studies with expressed UDP glucuronosyltransferases (UGTs) revealed that both UGT1A1 and UGT1A3 were capable of forming the glucuronide conjugates and the corresponding lactones for all three statins. Kinetic studies of statin glucuronidation and lactonization in liver microsomes revealed marked species differences in intrinsic clearance (CL(int)) values for SVA (but not for AVA or CVA), with the highest CL(int) observed in dogs, followed by rats and humans. Of the statins studied, SVA underwent glucuronidation and lactonization in human liver microsomes, with the lowest CL(int) (0.4 uL/min/mg of protein for SVA versus approximately 3 uL/min/mg of protein for AVA and CVA). Consistent with the present in vitro findings, substantial levels of the glucuronide conjugate (approximately 20% of dose) and the lactone form of SVA [simvastatin (SV); approximately 10% of dose] were detected in bile following i.v. administration of [(14)C]SVA to dogs. The acyl glucuronide conjugate of SVA, upon isolation from an in vitro incubation, underwent spontaneous cyclization to SV. Since the rate of this lactonization was high under conditions of physiological pH, the present results suggest that the statin lactones detected previously in bile and/or plasma following administration of SVA to animals or of AVA or CVA to animals and humans, might originate, at least in part, from the corresponding acyl glucuronide conjugates. Thus, acyl glucuronide formation, which seems to be a common metabolic pathway for the hydroxy acid forms of statins, may play an important, albeit previously unrecognized, role in the conversion of active HMG-CoA reductase inhibitors to their latent delta-lactone forms. The genetic variation underlying atorvastatin (ATV) pharmacokinetics was evaluated in a Mexican population. Aims of this study were: 1) to reveal the frequency of 87 polymorphisms in 36 genes related to drug metabolism in healthy Mexican volunteers, 2) to evaluate the impact of these polymorphisms on ATV pharmacokinetics, 3) to classify the ATV metabolic phenotypes of healthy volunteers, and 4) to investigate a possible association between genotypes and metabolizer phenotypes. A pharmacokinetic study of ATV (single 80-mg dose) was conducted in 60 healthy male volunteers. ATV plasma concentrations were measured by high-performance liquid chromatography mass spectrometry. Pharmacokinetic parameters were calculated by the non-compartmental method. The polymorphisms were determined with the PHARMAchip microarray and the TaqMan probes genotyping assay. Three metabolic phenotypes were found in our population: slow, normal, and rapid. Six gene polymorphisms were found to have a significant effect on ATV pharmacokinetics: MTHFR (rs1801133), DRD3 (rs6280), GSTM3 (rs1799735), TNFa (rs1800629), MDR1 (rs1045642), and SLCO1B1 (rs4149056). The combination of MTHFR, DRD3 and MDR1 polymorphisms associated with a slow ATV metabolizer phenotype. Atorvastatin has known human metabolites that include 7-[2-(4-Fluorophenyl)-4-[(4-hydroxyphenyl)carbamoyl]-3-phenyl-5-propan-2-ylpyrrol-1-yl]-3,5-dihydroxyheptanoic acid and 7-[2-(4-Fluorophenyl)-4-[(2-hydroxyphenyl)carbamoyl]-3-phenyl-5-propan-2-ylpyrrol-1-yl]-3,5-dihydroxyheptanoic acid. Atorvastatin is extensively metabolized to ortho- and parahydroxylated derivatives and various beta-oxidation products. In vitro inhibition of HMG-CoA reductase by ortho- and parahydroxylated metabolites is equivalent to that of atorvastatin. Approximately 70% of circulating inhibitory activity for HMG-CoA reductase is attributed to active metabolites. CYP3A4 is also involved in the metabolism of atorvastatin. Biological Half-Life The half-life of atorvastatin is 14 hours while the half-life of its metabolites can reach up to 30 hours. /MILK/ ...After administration to lactating rats, radioactivity in milk reached the maximum of 17.1 ng eq./mL at 6.0 hr and thereafter declined with a half-life of 7.8 hr. Mean plasma elimination half-life of Lipitor in humans is approximately 14 hours, but the half-life of inhibitory activity for HMG-CoA reductase is 20 to 30 hours due to the contribution of active metabolites. |
| Toxicity/Toxicokinetics |
Toxicity Summary
IDENTIFICATION AND USE: Atorvastatin is anticholesteremic agent and hydroxymethylglutaryl-CoA reductase inhibitor. HUMAN EXPOSURE AND TOXICITY: Cases of fatal and nonfatal hepatic failure have been reported rarely in patients receiving statins, including atorvastatin. Rhabdomyolysis with acute renal failure secondary to myoglobinuria also has been reported rarely in patients receiving statins, including atorvastatin. Lipid lowering drugs offer no benefit during pregnancy because cholesterol and cholesterol derivatives are needed for normal fetal development. Atherosclerosis is a chronic process, and discontinuation of lipid-lowering drugs during pregnancy should have little impact on long-term outcomes of primary hypercholesterolemia therapy. The occurrence of neuropsychiatric reactions is associated with statin treatment. They include behavioral alterations; cognitive and memory impairments; sleep disturbance; and sexual dysfunction. ANIMAL STUDIES: In a 2-year carcinogenicity study in rats at dose levels of 10, 30, and 100 mg/kg/day, 2 rare tumors were found in muscle in high-dose females: in one, there was a rhabdomyosarcoma, and in another, there was a fibrosarcoma. Atorvastatin caused no adverse effects on semen parameters, or reproductive organ histopathology in dogs given doses of 10, 40, or 120 mg/kg for two years. Male rats given 100 mg/kg/day for 11 weeks prior to mating had decreased sperm motility, spermatid head concentration, and increased abnormal sperm. Studies in rats performed at doses up to 175 mg/kg produced no changes in fertility. There was aplasia and aspermia in the epididymis of 2 of 10 rats treated with 100 mg/kg/day of atorvastatin for 3 months; testis weights were significantly lower at 30 and 100 mg/kg and epididymal weight was lower at 100 mg/kg. In a study in rats given 20, 100, or 225 mg/kg/day, from gestation day 7 through to lactation day 21 (weaning), there was decreased pup survival at birth, neonate, weaning, and maturity in pups of mothers dosed with 225 mg/kg/day. Body weight was decreased on days 4 and 21 in pups of mothers dosed at 100 mg/kg/day; pup body weight was decreased at birth and at days 4, 21, and 91 at 225 mg/kg/day. Pup development was delayed. In vitro, atorvastatin was not mutagenic or clastogenic in the following tests with and without metabolic activation: the Ames test with Salmonella typhimurium and Escherichia coli, the HGPRT forward mutation assay in Chinese hamster lung cells, and the chromosomal aberration assay in Chinese hamster lung cells. Atorvastatin was negative in the in vivo mouse micronucleus test. Atorvastatin selectively and competitively inhibits the hepatic enzyme HMG-CoA reductase. As HMG-CoA reductase is responsible for converting HMG-CoA to mevalonate in the cholesterol biosynthesis pathway, this results in a subsequent decrease in hepatic cholesterol levels. Decreased hepatic cholesterol levels stimulates upregulation of hepatic LDL-C receptors which increases hepatic uptake of LDL-C and reduces serum LDL-C concentrations. Toxicity Data Generally well-tolerated. Side effects may include myalgia, constipation, asthenia, abdominal pain, and nausea. Other possible side effects include myotoxicity (myopathy, myositis, rhabdomyolysis) and hepatotoxicity. To avoid toxicity in Asian patients, lower doses should be considered. Interactions Concomitant use of atorvastatin with efavirenz may result in reductions in plasma concentrations of atorvastatin. Following concomitant use of atorvastatin (10 mg daily for 3 days) and efavirenz (600 mg once daily for 14 days), atorvastatin peak plasma concentration and AUC were decreased by 1 and 41%, respectively. Concomitant use of atorvastatin (80 mg once daily for 14 days) and digoxin (0.25 mg once daily for 20 days) resulted in 20 and 15% increases in digoxin peak plasma concentration and AUC, respectively. Therefore, patients receiving such concomitant therapy should be monitored appropriately. Concomitant use of atorvastatin and azole antifungals (e.g., itraconazole) increases the risk of myopathy or rhabdomyolysis. Following concomitant use of atorvastatin (40 mg as a single dose) and itraconazole (200 mg once daily for 4 days), atorvastatin peak plasma concentration and area under the plasma concentration-time curve (AUC) were increased by 20% and 3.3-fold, respectively. Clinicians considering concomitant use of atorvastatin and itraconazole or other azole antifungals should weigh the benefits and risks of such concomitant therapy. During concomitant therapy with itraconazole, the lowest necessary dosage of atorvastatin should be employed, and dosage of atorvastatin should not exceed 20 mg daily. Patients receiving concomitant therapy with atorvastatin and azole antifungals should be monitored for manifestations of muscle pain, tenderness, or weakness, particularly during the initial months of therapy and following an increase in dosage of either drug. Concomitant use of atorvastatin and cyclosporine increases the risk of myopathy or rhabdomyolysis. Following concomitant use of atorvastatin (10 mg daily for 28 days) and cyclosporine (5.2 mg/kg daily), atorvastatin peak plasma concentration and AUC were increased by 10.7- and 8.7-fold, respectively. Concomitant use of atorvastatin and cyclosporine should be avoided. For more Interactions (Complete) data for ATORVASTATIN (27 total), please visit the HSDB record page. |
| References |
[1]. Santodomingo-Garzón T, et al. Atorvastatin inhibits inflammatory hypernociception. Br J Pharmacol. 2006 Sep;149(1):14-22.
[2]. Turner NA, et al. Comparison of the efficacies of five different statins on inhibition of human saphenous vein smooth muscle cell proliferation and invasion. J Cardiovasc Pharmacol. 2007 Oct;50(4):458-61. [3]. Nawrocki, J.W., et al., Reduction of LDL cholesterol by 25% to 60% in patients with primary hypercholesterolemia by atorvastatin, a new HMG-CoA reductase inhibitor. Arterioscler Thromb Vasc Biol, 1995. 15(5): p. 678-82. [4]. Song XJ, et al. Atorvastatin inhibits myocardial cell apoptosis in a rat model with post-myocardial infarction heart failure by downregulating ER stress response. Int J Med Sci. 2011;8(7):564-72. [5]. Li Y, et al. Inhibition of endoplasmic reticulum stress signaling pathway: A new mechanism of statins to suppress the development of abdominal aortic aneurysm. PLoS One. 2017 Apr 3;12(4):e0174821. [6]. Ming-Bai Hu, et al. Atorvastatin induces autophagy in MDA-MB-231 breast cancer cells. Ultrastruct Pathol. Sep-Oct 2018;42(5):409-415. [7]. In Vitro Screening for β-Hydroxy-β-methylglutaryl-CoA Reductase Inhibitory and Antioxidant Activity of Sequentially Extracted Fractions of Ficus palmata Forsk. Biomed Res Int. 2014; 2014: 762620. |
| Additional Infomation |
Therapeutic Uses
Anticholesteremic Agents; Hydroxymethylglutaryl-CoA Reductase Inhibitors In adult patients without clinically evident coronary heart disease, but with multiple risk factors for coronary heart disease such as age, smoking, hypertension, low HDL-C, or a family history of early coronary heart disease, Lipitor is indicated to: Reduce the risk of myocardial infarction; Reduce the risk of stroke; Reduce the risk for revascularization procedures and angina. /Included in US product label/ In patients with type 2 diabetes, and without clinically evident coronary heart disease, but with multiple risk factors for coronary heart disease such as retinopathy, albuminuria, smoking, or hypertension, Lipitor is indicated to: Reduce the risk of myocardial infarction; Reduce the risk of stroke. /Included in US product label/ In patients with clinically evident coronary heart disease, Lipitor is indicated to: Reduce the risk of non-fatal myocardial infarction; Reduce the risk of fatal and non-fatal stroke; Reduce the risk for revascularization procedures; Reduce the risk of hospitalization for congestive heart failure (CHF); Reduce the risk of angina. /Included in US product label/ For more Therapeutic Uses (Complete) data for ATORVASTATIN (15 total), please visit the HSDB record page. Drug Warnings Lipitor is contraindicated in women who are or may become pregnant. Serum cholesterol and triglycerides increase during normal pregnancy. Lipid lowering drugs offer no benefit during pregnancy because cholesterol and cholesterol derivatives are needed for normal fetal development. Atherosclerosis is a chronic process, and discontinuation of lipid-lowering drugs during pregnancy should have little impact on long-term outcomes of primary hypercholesterolemia therapy. Statins may cause fetal harm when administered to a pregnant woman. Lipitor should be administered to women of childbearing potential only when such patients are highly unlikely to conceive and have been informed of the potential hazards. If the woman becomes pregnant while taking Lipitor, it should be discontinued immediately and the patient advised again as to the potential hazards to the fetus and the lack of known clinical benefit with continued use during pregnancy. It is not known whether atorvastatin is excreted in human milk, but a small amount of another drug in this class does pass into breast milk. Nursing rat pups had plasma and liver drug levels of 50% and 40%, respectively, of that in their mother's milk. Animal breast milk drug levels may not accurately reflect human breast milk levels. Because another drug in this class passes into human milk and because statins have a potential to cause serious adverse reactions in nursing infants, women requiring Lipitor treatment should be advised not to nurse their infants. Myopathy (defined as muscle aches or weakness in conjunction with increases in creatine kinase [CK, creatine phosphokinase, CPK] concentrations exceeding 10 times the upper limit of normal [ULN]) has been reported occasionally in patients receiving statins, including atorvastatin. Rhabdomyolysis with acute renal failure secondary to myoglobinuria also has been reported rarely in patients receiving statins, including atorvastatin. For more Drug Warnings (Complete) data for ATORVASTATIN (33 total), please visit the HSDB record page. Pharmacodynamics Atorvastatin is an oral antilipemic agent that reversibly inhibits HMG-CoA reductase. It lowers total cholesterol, low-density lipoprotein-cholesterol (LDL-C), apolipoprotein B (apo B), non-high density lipoprotein-cholesterol (non-HDL-C), and triglyceride (TG) plasma concentrations while increasing HDL-C concentrations. High LDL-C, low HDL-C and high TG concentrations in the plasma are associated with increased risk of atherosclerosis and cardiovascular disease. The total cholesterol to HDL-C ratio is a strong predictor of coronary artery disease, and high ratios are associated with a higher risk of disease. Increased levels of HDL-C are associated with lower cardiovascular risk. By decreasing LDL-C and TG and increasing HDL-C, atorvastatin reduces the risk of cardiovascular morbidity and mortality. Elevated cholesterol levels (and high low-density lipoprotein (LDL) levels in particular) are an important risk factor for the development of CVD. Clinical studies have shown that atorvastatin reduces LDL-C and total cholesterol by 36-53%. In patients with dysbetalipoproteinemia, atorvastatin reduced the levels of intermediate-density lipoprotein cholesterol. It has also been suggested that atorvastatin can limit the extent of angiogenesis, which can be useful in the treatment of chronic subdural hematoma. **Myopathy/Rhabdomyolysis** Atorvastatin, like other HMG-CoA reductase inhibitors, is associated with a risk of drug-induced myopathy characterized by muscle pain, tenderness, or weakness in conjunction with elevated levels of creatine kinase (CK). Myopathy often manifests as rhabdomyolysis with or without acute renal failure secondary to myoglobinuria. The risk of statin-induced myopathy is dose-related, and the symptoms of myopathy are typically resolved upon drug discontinuation. Results from observational studies suggest that 10-15% of people taking statins may experience muscle aches at some point during treatment. **Liver Dysfunction** Statins, like some other lipid-lowering therapies, have been associated with biochemical abnormalities of liver function. Persistent elevations (> 3 times the upper limit of normal [ULN] occurring on two or more occasions) in serum transaminases occurred in 0.7% of patients who received atorvastatin in clinical trials. This effect appears to be dose-related. **Endocrine Effects** Statins are associated with a risk of increased serum HbA1c and glucose levels. An _in vitro_ study demonstrated a dose-dependent cytotoxic effect on human pancreatic islet β cells following treatment with atorvastatin. Moreover, insulin secretion rates decreased relative to control. HMG-CoA reductase inhibitors interfere with cholesterol synthesis and may theoretically interfere with the production of adrenal and/or gonadal steroids. Clinical studies with atorvastatin and other HMG-CoA reductase inhibitors have suggested that these agents do not affect plasma cortisol concentrations, basal plasma testosterone concentration, or adrenal reserve. However, the effect of statins on male fertility has not been fully investigated. The effects of statins on the pituitary-gonadal axis in premenopausal women are unknown. **Cardiovascular** Significant decreases in circulating ubiquinone levels in patients treated with atorvastatin and other statins have been observed. The clinical significance of a potential long-term statin-induced deficiency of ubiquinone has not been established. It has been reported that a decrease in myocardial ubiquinone levels could lead to impaired cardiac function in patients with borderline congestive heart failure. **Lipoprotein A** In some patients, the beneficial effect of lowered total cholesterol and LDL-C levels may be partly blunted by the concomitant increase in Lp(a) lipoprotein concentrations. Present knowledge suggests the importance of high Lp(a) levels as an emerging risk factor for coronary heart disease. Further studies have demonstrated statins affect Lp(a) levels differently in patients with dyslipidemia depending on their apo(a) phenotype; statins increase Lp(a) levels exclusively in patients with the low molecular weight apo(a) phenotype. |
| Molecular Formula |
C33H35FN2O5
|
|---|---|
| Molecular Weight |
558.65
|
| Exact Mass |
558.253
|
| Elemental Analysis |
C, 70.95; H, 6.32; F, 3.40; N, 5.01; O, 14.32
|
| CAS # |
134523-00-5
|
| Related CAS # |
Atorvastatin hemicalcium salt;134523-03-8;(3S,5S)-Atorvastatin;501121-34-2;Atorvastatin-d5 hemicalcium;222412-82-0;(rel)-Atorvastatin;110862-48-1;Atorvastatin hemicalcium trihydrate;344920-08-7;Atorvastatin-d5 sodium;222412-87-5; 609843-23-4 (lysine);
344423-98-9 (calcium trihydrate); 1035609-19-8 (magnesium trihydrate); 134523-00-5 (free acid); 1072903-92-4 (strontium) ; 134523-01-6 (sodium); 874114-41-7 (magnesium);
|
| PubChem CID |
60823
|
| Appearance |
White to light yellow solid powder
|
| Density |
1.2±0.1 g/cm3
|
| Boiling Point |
722.2±60.0 °C at 760 mmHg
|
| Melting Point |
176-178°C
|
| Flash Point |
390.6±32.9 °C
|
| Vapour Pressure |
0.0±2.5 mmHg at 25°C
|
| Index of Refraction |
1.603
|
| LogP |
4.13
|
| Hydrogen Bond Donor Count |
4
|
| Hydrogen Bond Acceptor Count |
6
|
| Rotatable Bond Count |
12
|
| Heavy Atom Count |
41
|
| Complexity |
822
|
| Defined Atom Stereocenter Count |
2
|
| SMILES |
FC1C([H])=C([H])C(=C([H])C=1[H])C1=C(C2C([H])=C([H])C([H])=C([H])C=2[H])C(C(N([H])C2C([H])=C([H])C([H])=C([H])C=2[H])=O)=C(C([H])(C([H])([H])[H])C([H])([H])[H])N1C([H])([H])C([H])([H])[C@]([H])(C([H])([H])[C@]([H])(C([H])([H])C(=O)O[H])O[H])O[H]
|
| InChi Key |
XUKUURHRXDUEBC-KAYWLYCHSA-N
|
| InChi Code |
InChI=1S/C33H35FN2O5/c1-21(2)31-30(33(41)35-25-11-7-4-8-12-25)29(22-9-5-3-6-10-22)32(23-13-15-24(34)16-14-23)36(31)18-17-26(37)19-27(38)20-28(39)40/h3-16,21,26-27,37-38H,17-20H2,1-2H3,(H,35,41)(H,39,40)/t26-,27-/m1/s1
|
| Chemical Name |
(3R,5R)-7-(2-(4-fluorophenyl)-5-isopropyl-3-phenyl-4-(phenylcarbamoyl)-1H-pyrrol-1-yl)-3,5-dihydroxyheptanoic acid
|
| Synonyms |
CI-981; CI981; Atorvastatin; liptonorm; Lipilou; Tozalip; Xavator; Lipitor; Cardyl; ATORVASTATIN CALCIUM; Torvast; Cardyl
|
| HS Tariff Code |
2934.99.9001
|
| Storage |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| Solubility (In Vitro) |
DMSO : ~50 mg/mL (~89.50 mM)
|
|---|---|
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.5 mg/mL (4.48 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution.
For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2.5 mg/mL (4.48 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.7900 mL | 8.9501 mL | 17.9003 mL | |
| 5 mM | 0.3580 mL | 1.7900 mL | 3.5801 mL | |
| 10 mM | 0.1790 mL | 0.8950 mL | 1.7900 mL |
*Note: Please select an appropriate solvent for the preparation of stock solution based on your experiment needs. For most products, DMSO can be used for preparing stock solutions (e.g. 5 mM, 10 mM, or 20 mM concentration); some products with high aqueous solubility may be dissolved in water directly. Solubility information is available at the above Solubility Data section. Once the stock solution is prepared, aliquot it to routine usage volumes and store at -20°C or -80°C. Avoid repeated freeze and thaw cycles.
Calculation results
Working concentration: mg/mL;
Method for preparing DMSO stock solution: mg drug pre-dissolved in μL DMSO (stock solution concentration mg/mL). Please contact us first if the concentration exceeds the DMSO solubility of the batch of drug.
Method for preparing in vivo formulation::Take μL DMSO stock solution, next add μL PEG300, mix and clarify, next addμL Tween 80, mix and clarify, next add μL ddH2O,mix and clarify.
(1) Please be sure that the solution is clear before the addition of next solvent. Dissolution methods like vortex, ultrasound or warming and heat may be used to aid dissolving.
(2) Be sure to add the solvent(s) in order.
|
|
|
Products are for research use only; We do not sell to patients
Copyright 2020 InvivoChem LLC | All Rights Reserved



























 COA
COA