| Size | Price | Stock | Qty |
|---|---|---|---|
| 2mg |
|
||
| 5mg |
|
||
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| Other Sizes |
Purity: =98.1%
PLX8394 (PLX-8394) is a novel, potent, orally bioavailable and selective inhibitor of the serine/threonine-protein kinase B-raf (BRaf) with anticancer activity. With an approximate 5 nM IC50 for BRAFV600E, it inhibits BRAF. In BRAF-mutant LA models, PLX8394 can avoid the reactivation of the MAPK pathway. Treatment with PLX8394 is successful in vitro and in vivo in BRAFV600E and some non-V600 LA models. Treatment-naive BRAF-mutant LAs and those with acquired vemurafenib resistance brought on by an alternatively spliced, truncated BRAFV600E that encourages vemurafenib-insensitive MAPK pathway signaling were both successfully treated with PLX8394. A combination therapy that includes PLX8394 and either an EGFR or mTOR inhibitor up front can prevent acquired PLX8394 resistance, which is caused by EGFR-mediated RAS-mTOR signaling. This study offers a biological justification and potential polytherapy plan to support the administration of PLX8394 to patients with lung cancer. By encouraging mitogen-activated protein kinase (MAPK) pathway signaling, oncogenic activation of the protein kinase BRAF promotes tumor growth. BRAF-mutant lung adenocarcinoma (LA) is the most common cause of BRAF-mutant cancer mortality globally because oncogenic mutations in BRAF occur in ~2-7% of LA. However, the spectrum of BRAF mutations in LA includes both BRAFV600E (60% of cases) and non-V600E mutant alleles (~40% of cases), such as BRAFG469A and BRAFG466V. Unlike most tumor types, which primarily harbor the BRAFV600E-mutant allele, LA tumors primarily harbor the BRAFV600E-mutant allele. Clinical trials testing selective BRAF inhibitors, like vemurafenib, in BRAFV600E-mutant patients have been prompted by the presence of BRAFV600E in LA. Despite showing some clinical promise, reactivation of the MAPK pathway signaling frequently leads to both innate and acquired resistance, which limits the long-lasting effects of the available BRAF inhibitors. Furthermore, the ideal therapeutic approach to stop non-V600E BRAF-mutant LA is still unknown.
| Targets |
BRAF(V600E) (IC50 = 3.8 nM); BRAF (IC50 = 14 nM); CRAF (IC50 = 23 nM)
|
|---|---|
| ln Vitro |
PLX8394 is a next-generation, orally available small-molecule BRAFi that inhibits both monomeric and dimeric BRAFV600 and BRAFnon-V600 protein signaling and does not cause the RAF/MEK/ERK paradoxical activation[1].
Unlike first-generation BRAFi, PLX8394 [a] does not induce paradoxical activation of the RAF/MEK/ERK pathway in cells with stimulated RAS signaling, and [b] blocks signaling from both monomeric BRAFV600 and dimeric BRAFnon-V600 mutations.[1] PLX8394, a new generation BRAF inhibitor, selectively inhibits BRAF in colonic adenocarcinoma cells and prevents paradoxical MAPK pathway activation. [3] PLX8394 overcame RAF inhibitor resistance in BRAF fusions characterizing paediatric astrocytomas28 and maintained activity against cells that are vemurafenib-resistant through secondary mutation in NRAS. [2] |
| ln Vivo |
PLX8394 is a small molecule BRAF inhibitor of the newest generation that can be taken orally and has antineoplastic potential.
Of 26 patients who received at least 1 dose of PLX8394, 15 patients (10 with BRAF mutation) were enrolled to cohorts with PLX8394 monotherapy: 450mg BID (n=8), 900mg BID (n=3), 450mg TID (n=4). PK studies demonstrated lower than expected drug exposures; the protocol was amended and 11 additional patients (8 with BRAF mutation) were enrolled in cohorts with PLX8394 coadministered with cobicistat: 450mg BID (n=5) and 900mg BID (n=6). As of 31 May 2017 cutoff, 15 subjects terminated the study: 14 for disease progression, 1 subject withdrawal. Duration of study drug exposure was a median of 56 (22 - 727) days. Reversible Grade (G) 3 transaminitis in a patient treated with PLX8394 900mg BID + cobicistat was the only dose-limiting toxicity (DLT); rechallenge with PLX8394 alone was tolerated without recurrence of transaminitis. PLX8394 900mg BID + cobicistat was declared as RP2D. Treatment-related G>3 AEs other than DLT included G3 diarrhea (n=1), G3 anorexia (n=1), both in PLX8394 monotherapy cohorts and both manageable with supportive care. There were no cutaneous or ocular AEs. Cobicistat coadministration resulted in a 2-3 fold increase in PLX8394 systemic exposure, with RP2D achieving the projected efficacious exposure range based on preclinical models. There were no objective responses in PLX8394 monotherapy cohorts; however, to date 2 (28%) of 7 evaluable BRAF mutant patients treated with PLX8394 + cobicistat achieved partial responses (BRAFV600E-mutated colorectal cancer, 42% and BRAFV600E-mutated glioma, 65%; both BRAFi naive), which were confirmed by 8-wk serial scans. Tumors without BRAF mutation showed no response. Conclusion: PLX8394 with cobicistat was very well tolerated and showed promising activity in refractory solid tumors with BRAF mutation. A phase II study in BRAF-mutated cancers is ongoing. [1] To construct therapeutic strategies for treating cancers harboring LLRins506 or VLRins506 mutation, we next determined the efficacies of RAF inhibitors (vemurafenib, dabrafenib, and PLX8394) and MEK inhibitor (trametinib) against fibroblastomas induced by LLRins506 or VLRins506 mutant. Although both RAF inhibitors and MEK inhibitor impaired the growth of BRAF(V600E)–induced fibroblastomas by different extends (trametinib > dabrafenib > vemurafenib > PLX8394), only MEK inhibitor exhibited a strong inhibitory effect on that of LLRins506/VLRins506 mutant–induced fibroblastomas (Fig. 6, D to F). Consistent with this finding, our immunohistochemical staining revealed that administrating with MEK inhibitor, trametinib, but not with RAF inhibitors significantly decreased the level of phospho-ERK1/2 and Ki67 in LLRins506/VLRins506 mutant–induced fibroblastomas (Fig. 6G). Together, these data demonstrated that LLRins506 and VLRins506 are real driver mutations in cancer genomes and cancers harboring this type of mutations could be treated effectively with MEK inhibitor, trametinib.[4] |
| Enzyme Assay |
For in vitro kinase assays of BRAF mutants, the immunoprecipitants were washed once with kinase reaction buffer (25 mM Hepes, 10 mM MgCl2, 0.5 mM Na3VO4, 0.5 mM dithiothreitol, pH 7.4) and then incubated with 20-μl kinase reaction mixture [2-μg His-tagged MEK1(K97A) and 100 μM adenosine 5′-triphosphate in 20-μl kinase reaction buffer] per sample at room temperature for 10 min. Kinase reaction was stopped by adding 5 μl per sample of 5× Laemmli sample buffer and determined by immunoblotting. Otherwise, the immunoprecipitants were directly mixed with 2× Laemmli sample buffer before detection. The immunoblotting was carried out as described before [4].
MST analysis [4] The affinity of BRAF(V600E) and LLRins506 mutants with different RAF inhibitors (vemurafenib, dabrafenib, and PLX8394) was measured using the Monolith NT.115 from NanoTemper Technologies. The MST analysis was carried out as described before. Briefly, BRAF mutants purified from 293T transfectants were labeled with a fluorescent dye NT-647 (Cysteine Reactive) according to the manufacturer’s protocol. Then, a series of protein solutions with different concentrations were prepared by consecutive twofold dilutions in buffer containing 25 mM Hepes (pH 7.5), 150 mM NaCl, 0.2% IGEPAL, and 0.1 mM Tris(2-carboxyethyl)Phosphine (TCEP). The labeled proteins were mixed with unlabeled drugs at a volume ratio of 1:1 and loaded into silica capillaries after a short incubation at room temperature. The measurements were performed at 25°C by using 20% light-emitting diode power and 40% MST power. The laser-on and laser-off intervals were 30 and 5 s, respectively. NanoTemper Analysis (x86) software was used to fit the data and to determine the apparent dissociation constant values. |
| Cell Assay |
Cellular proliferation assay [3]
Cellular proliferation assay of colon cancer cell lines was performed on Falcon® 96 well clear plates. Cells were seeded at 3000 cells per well. Cells were incubated with the inhibitors vemurafenib or PLX8394 at concentrations of 0 µM (DMSO), 0.1 µM, 0.5 µM, and 1 µM. Proliferation assays were performed using the MTS-based CellTiter 96® AQueous One Solution Cell Proliferation Assay. Briefly, cells were incubated for 1 hour in diluted CellTiter mixture (1:5 dilution in growth media) prior to measurement of absorbance at 490nm wavelength and subtraction of background as measured from no-cell control wells. Readings were performed at day 1 before treatment and after 72 hours (3 days) of inhibitor treatment. Absorbance were normalised within cell lines to untreated controls. [3] |
| Animal Protocol |
For xenograft experiments, female NOD-SCID mice (6 to 8 weeks) were subcutaneously injected with 5 × 106 cells per mice in 1:1 Matrigel. Tumor volumes were monitored by digital calipers twice a week and calculated using the formula: volume = (width)2 × length/2. Vemurafenib (120 mg/kg), dabrafenib (200 mg/kg), PLX8394 (150 mg/kg), and trametinib (3 mg/kg) were administered orally and daily when tumors reached an average volume of ~100 mm3 according to the previous studies. At the experiment endpoint, mice were euthanized, and tumors were harvested for ex vivo analysis and subsequent histology. All operations were approved by the Animal Ethics Committee of National Cancer Centre Singapore (NCCS) with Institutional Animal Care and Use Committee (IACUC)–approved animal protocol. [4]
Trial protocol PLX120-03 (NCT02428712) incorporates an open-label, multicenter, phase I study using a 3+3 design to determine the recommended phase II dose (RP2D) in patients with refractory or relapsed solid tumors with or without BRAF mutation. Other endpoints are safety, including dermatologic and ophthalmic adverse events (AEs), pharmacokinetics (PK), and response per RECIST1.1. Dose escalation includes cohorts with either PLX8394 monotherapy or PLX8394 coadministered with an oral CYP3A4 inhibitor, cobicistat 150mg, to enhance systemic PK. Enrollment at the RP2D continues in the subsequent phase II study.[1] |
| References | |
| Additional Infomation |
PLX8394 is under investigation in clinical trial NCT02428712 (A Study of PLX8394 as a Single Agent in Patients With Advanced Unresectable Solid Tumors).
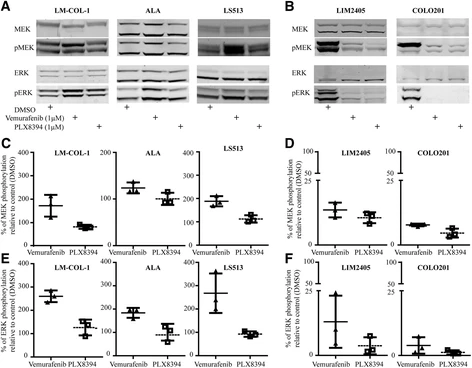
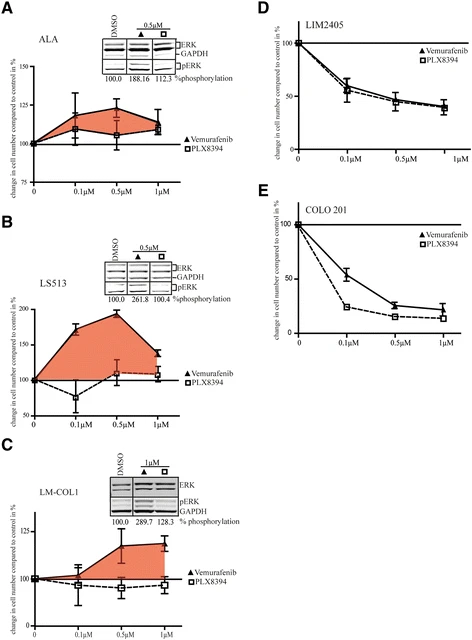
Plixorafenib is an orally bioavailable inhibitor and specific dimer breaker of the serine/threonine-protein kinase B-raf (BRAF) protein, with potential antineoplastic activity. Upon oral administration, plixorafenib selectively binds to and inhibits the activity of dimeric BRAF mutants, including BRAF fusions and splice variants, and BRAFV600 monomers, while sparing RAF function in normal cells. This inhibits the proliferation of tumor cells which express these mutated forms of BRAF. BRAF, a member of the raf family of serine/threonine protein kinases, plays a role in the regulation of mitogen-activated protein kinase (MAPK) and extracellular signal-regulated kinase (ERK) signaling pathways, which may be constitutively activated due to BRAF gene mutations. Mutated forms and fusions of BRAF are associated with a number of neoplastic diseases. Oncogenic activation of BRAF fuels cancer growth by constitutively promoting RAS-independent mitogen-activated protein kinase (MAPK) pathway signalling. Accordingly, RAF inhibitors have brought substantially improved personalized treatment of metastatic melanoma. However, these targeted agents have also revealed an unexpected consequence: stimulated growth of certain cancers. Structurally diverse ATP-competitive RAF inhibitors can either inhibit or paradoxically activate the MAPK pathway, depending whether activation is by BRAF mutation or by an upstream event, such as RAS mutation or receptor tyrosine kinase activation. Here we have identified next-generation RAF inhibitors (dubbed 'paradox breakers') that suppress mutant BRAF cells without activating the MAPK pathway in cells bearing upstream activation. In cells that express the same HRAS mutation prevalent in squamous tumours from patients treated with RAF inhibitors, the first-generation RAF inhibitor vemurafenib stimulated in vitro and in vivo growth and induced expression of MAPK pathway response genes; by contrast the paradox breakers PLX7904 and PLX8394 had no effect. Paradox breakers also overcame several known mechanisms of resistance to first-generation RAF inhibitors. Dissociating MAPK pathway inhibition from paradoxical activation might yield both improved safety and more durable efficacy than first-generation RAF inhibitors, a concept currently undergoing human clinical evaluation with PLX8394.[2] BRAF inhibitors (BRAFi) are standard of care for the treatment of BRAF V600 mutation-driven metastatic melanoma, but can lead to paradoxical activation of the mitogen-activated protein kinase (MAPK) signalling pathway. This can result in the promotion of precancerous lesions and secondary neoplasms, mainly (but not exclusively) associated with pre-existing mutations in RAS genes. We previously reported a patient with synchronous BRAF-mutated metastatic melanoma and BRAF wt /KRAS G12D-metastatic colorectal cancer (CRC), whose CRC relapsed and progressed when treated with the BRAF inhibitor dabrafenib (GSK2118436). We used tissue from the resected CRC metastasis to derive a cell line, LM-COL-1, which directly and reliably mimicked the clinical scenario including paradoxical activation of the MAPK signalling pathway resulting in increased cell proliferation upon dabrafenib treatment. Novel BRAF inhibitors (PLX8394 and PLX7904), dubbed as "paradox breakers", were developed to inhibit V600 mutated oncogenic BRAF without causing paradoxical MAPK pathway activation. In this study we used our LM-COL-1 model alongside multiple other CRC cell lines with varying mutational backgrounds to demonstrate and confirm that the paradox breaker PLX8394 retains on-target inhibition of mutated BRAF V600 without paradoxically promoting MAPK signalling.[3] Although targeting BRAF mutants with RAF inhibitors has achieved promising outcomes in cancer therapy, drug resistance remains a remarkable challenge, and underlying molecular mechanisms are not fully understood. Here, we characterized a previously unknown group of oncogenic BRAF mutants with in-frame insertions (LLRins506 or VLRins506) of αC-β4 loop. Using structure modeling and molecular dynamics simulation, we found that these insertions formed a large hydrophobic network that stabilizes R-spine and thus triggers the catalytic activity of BRAF. Furthermore, these insertions disrupted BRAF dimer interface and impaired dimerization. Unlike BRAF(V600E), these BRAF mutants with low dimer affinity were strongly resistant to all RAF inhibitors in clinic or clinical trials, which arises from their stabilized R-spines. As predicted by molecular docking, the stabilized R-spines in other BRAF mutants also conferred drug resistance. Together, our data indicated that the stability of R-spine but not dimer affinity determines the RAF inhibitor resistance of oncogenic BRAF mutants.[4] |
| Molecular Formula |
C25H21F3N6O3S
|
|---|---|
| Molecular Weight |
542.5329
|
| Exact Mass |
542.134
|
| Elemental Analysis |
C, 55.35; H, 3.90; F, 10.51; N, 15.49; O, 8.85; S, 5.91
|
| CAS # |
1393466-87-9
|
| Related CAS # |
1393466-87-9 (PLX8394); 1652573-86-8 (PLX7683, a paradox breaker); 918505-61-0 (Vemurafenib/ PLX4032 analog)
|
| PubChem CID |
90116675
|
| Appearance |
White to off-white solid powder
|
| Density |
1.6±0.1 g/cm3
|
| Boiling Point |
786.8±70.0 °C at 760 mmHg
|
| Flash Point |
429.6±35.7 °C
|
| Vapour Pressure |
0.0±2.7 mmHg at 25°C
|
| Index of Refraction |
1.703
|
| LogP |
0.97
|
| Hydrogen Bond Donor Count |
2
|
| Hydrogen Bond Acceptor Count |
11
|
| Rotatable Bond Count |
7
|
| Heavy Atom Count |
38
|
| Complexity |
976
|
| Defined Atom Stereocenter Count |
1
|
| SMILES |
S(N([H])C1C([H])=C([H])C(=C(C(C2=C([H])N([H])C3=C2C([H])=C(C([H])=N3)C2C([H])=NC(C3([H])C([H])([H])C3([H])[H])=NC=2[H])=O)C=1F)F)(N1C([H])([H])C([H])([H])[C@]([H])(C1([H])[H])F)(=O)=O
|
| InChi Key |
YYACLQUDUDXAPA-MRXNPFEDSA-N
|
| InChi Code |
InChI=1S/C25H21F3N6O3S/c26-16-5-6-34(12-16)38(36,37)33-20-4-3-19(27)21(22(20)28)23(35)18-11-32-25-17(18)7-14(8-31-25)15-9-29-24(30-10-15)13-1-2-13/h3-4,7-11,13,16,33H,1-2,5-6,12H2,(H,31,32)/t16-/m1/s1
|
| Chemical Name |
(3R)-N-[3-[5-(2-cyclopropylpyrimidin-5-yl)-1H-pyrrolo[2,3-b]pyridine-3-carbonyl]-2,4-difluorophenyl]-3-fluoropyrrolidine-1-sulfonamide
|
| Synonyms |
PLX-8394; PLX 8394; PLX8394; 1393466-87-9; Plixorafenib; (R)-N-(3-(5-(2-cyclopropylpyrimidin-5-yl)-1H-pyrrolo[2,3-b]pyridine-3-carbonyl)-2,4-difluorophenyl)-3-fluoropyrrolidine-1-sulfonamide; UNII-J2L7Z273SG; FORE8394; PLX8394
|
| HS Tariff Code |
2934.99.9001
|
| Storage |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| Solubility (In Vitro) |
DMSO: ~100 mg/mL (~184.3 mM)
|
|---|---|
| Solubility (In Vivo) |
Note: Listed below are some common formulations that may be used to formulate products with low water solubility (e.g. < 1 mg/mL), you may test these formulations using a minute amount of products to avoid loss of samples.
Injection Formulations
Injection Formulation 1: DMSO : Tween 80: Saline = 10 : 5 : 85 (i.e. 100 μL DMSO stock solution → 50 μL Tween 80 → 850 μL Saline)(e.g. IP/IV/IM/SC) *Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH ₂ O to obtain a clear solution. Injection Formulation 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL DMSO → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Injection Formulation 3: DMSO : Corn oil = 10 : 90 (i.e. 100 μL DMSO → 900 μL Corn oil) Example: Take the Injection Formulation 3 (DMSO : Corn oil = 10 : 90) as an example, if 1 mL of 2.5 mg/mL working solution is to be prepared, you can take 100 μL 25 mg/mL DMSO stock solution and add to 900 μL corn oil, mix well to obtain a clear or suspension solution (2.5 mg/mL, ready for use in animals). View More
Injection Formulation 4: DMSO : 20% SBE-β-CD in saline = 10 : 90 [i.e. 100 μL DMSO → 900 μL (20% SBE-β-CD in saline)] Oral Formulations
Oral Formulation 1: Suspend in 0.5% CMC Na (carboxymethylcellulose sodium) Oral Formulation 2: Suspend in 0.5% Carboxymethyl cellulose Example: Take the Oral Formulation 1 (Suspend in 0.5% CMC Na) as an example, if 100 mL of 2.5 mg/mL working solution is to be prepared, you can first prepare 0.5% CMC Na solution by measuring 0.5 g CMC Na and dissolve it in 100 mL ddH2O to obtain a clear solution; then add 250 mg of the product to 100 mL 0.5% CMC Na solution, to make the suspension solution (2.5 mg/mL, ready for use in animals). View More
Oral Formulation 3: Dissolved in PEG400 (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.8432 mL | 9.2161 mL | 18.4322 mL | |
| 5 mM | 0.3686 mL | 1.8432 mL | 3.6864 mL | |
| 10 mM | 0.1843 mL | 0.9216 mL | 1.8432 mL |
*Note: Please select an appropriate solvent for the preparation of stock solution based on your experiment needs. For most products, DMSO can be used for preparing stock solutions (e.g. 5 mM, 10 mM, or 20 mM concentration); some products with high aqueous solubility may be dissolved in water directly. Solubility information is available at the above Solubility Data section. Once the stock solution is prepared, aliquot it to routine usage volumes and store at -20°C or -80°C. Avoid repeated freeze and thaw cycles.
Calculation results
Working concentration: mg/mL;
Method for preparing DMSO stock solution: mg drug pre-dissolved in μL DMSO (stock solution concentration mg/mL). Please contact us first if the concentration exceeds the DMSO solubility of the batch of drug.
Method for preparing in vivo formulation::Take μL DMSO stock solution, next add μL PEG300, mix and clarify, next addμL Tween 80, mix and clarify, next add μL ddH2O,mix and clarify.
(1) Please be sure that the solution is clear before the addition of next solvent. Dissolution methods like vortex, ultrasound or warming and heat may be used to aid dissolving.
(2) Be sure to add the solvent(s) in order.
| NCT Number | Recruitment | interventions | Conditions | Sponsor/Collaborators | Start Date | Phases |
| NCT02012231 | Terminated | Drug: PLX8394 | Melanoma Histiocytosis |
Fore Biotherapeutics | February 2014 | Phase 1 |
|
|
Products are for research use only; We do not sell to patients
Copyright 2020 InvivoChem LLC | All Rights Reserved



























































 COA
COA