| Size | Price | Stock | Qty |
|---|---|---|---|
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| 500mg |
|
||
| Other Sizes |
|
































| Targets |
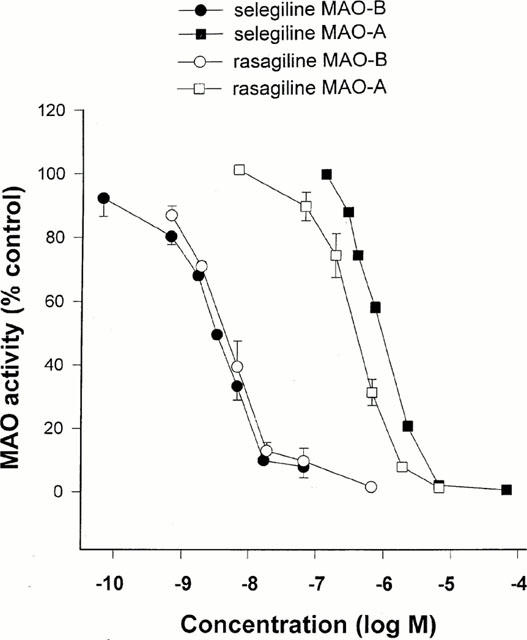
rMAO-B (IC50 = 4.43 nM); rMAO-A (IC50 = 412 nM)
|
|---|---|
| ln Vitro |
Following treatment with dexamethasone (10 µM), the proliferation rate of SH-SY5Y and 1242-MG was considerably boosted by rasagine (0.25 nM; 96 hours) [2].
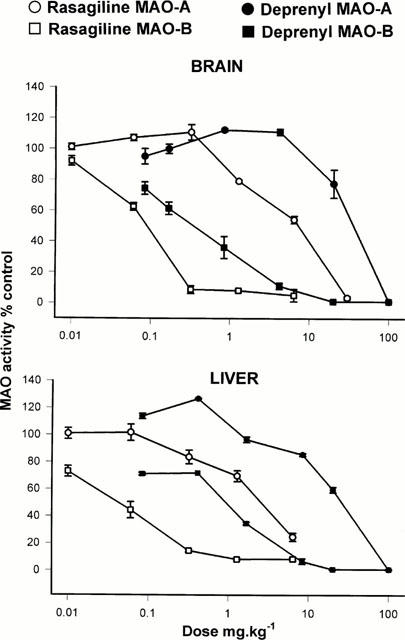
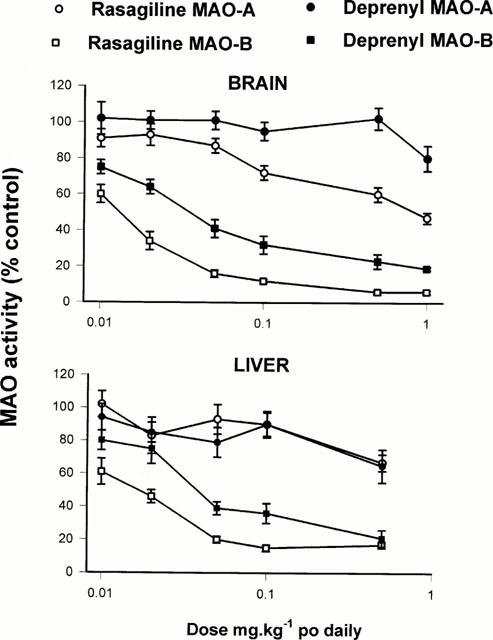
1. Rasagiline [N-propargyl-1R(+)-aminoindan], was examined for its monoamine oxidase (MAO) A and B inhibitor activities in rats together with its S(-)-enantiomer (TVP 1022) and the racemic compound (AGN-1135) and compared to selegiline (1-deprenyl). The tissues that were studied for MAO inhibition were the brain, liver and small intestine. 2. While rasagiline and AGN1135 are highly potent selective irreversible inhibitors of MAO in vitro and in vivo, the S(-) enantiomer is relatively inactive in the tissues examined. 3. The in vitro IC(50) values for inhibition of rat brain MAO activity by rasagiline are 4.43+/-0.92 nM (type B), and 412+/-123 nM (type A). The ED(50) values for ex vivo inhibition of MAO in the brain and liver by a single dose of rasagiline are 0.1+/-0.01, 0.042+/-0.0045 mg kg(-1) respectively for MAO-B, and 6.48+/-0.81, 2.38+/-0.35 mg kg(-1) respectively for MAO-A. 4. Selective MAO-B inhibition in the liver and brain was maintained on chronic (21 days) oral dosage with ED(50) values of 0.014+/-0.002 and 0.013+/-0.001 mg kg(-1) respectively. 5. The degree of selectivity of rasagiline for inhibition of MAO-B as opposed to MAO-A was similar to that of selegiline. Rasagiline was three to 15 times more potent than selegiline for inhibition of MAO-B in rat brain and liver in vivo on acute and chronic administration, but had similar potency in vitro. 6. These data together with lack of tyramine sympathomimetic potentiation by rasagiline, at selective MAO-B inhibitory dosage, indicate that this inhibitor like selegiline may be a useful agent in the treatment of Parkinson's disease in either symptomatic or L-DOPA adjunct therapy, but lack of amphetamine-like metabolites could present a therapeutic advantage for rasagiline.[1] Stress can affect the brain and lead to depression; however, the molecular pathogenesis is unclear. An association between stress and stress-induced hypersecretion of glucocorticoids occurs during stress. Dexamethasone (a synthetic glucocorticoid steroid) has been reported to induce apoptosis and increase the activity of monoamine oxidase (MAO) (Youdim et al. 1989). MAO is an enzyme for the degradation of aminergic neurotransmitters; dopamine, noradrenaline and serotonin and dietary amines and MAO inhibitors are classical antidepressant drugs. In this study, we have compared the ability of rasagiline (Azilect) and its main metabolite, R-aminoindan with selegiline (Deprenyl) in prevention of dexamethasone-induced brain cell death employing human neuroblastoma SH-SY5Y cells and glioblastoma 1242-MG cells. Dexamethasone reduced cell viability as measured by MTT test, but rasagiline, selegiline, and 1-R-aminoindan could significantly prevent dexamethasone-induced brain cell death. Among three drugs, rasagiline had the highest neuroprotective effect. Furthermore, the inhibitory effects of these drugs on MAO B catalytic activity and on apoptotic DNA damage (TUNEL staining) were examined. Rasagiline exhibited highest inhibition on MAO B enzymatic activity and prevention on DNA damage as compared to selegiline and 1-R-aminoindan. In summary, the greater neuroprotective effect of rasagiline may be associated with the combination of the parent drug and its metabolite 1-R-aminoindan.[2] |
| ln Vivo |
Under a transgenic model of multiple system atrophy, rasagiline is neuroprotective. Treatment with 2.5 mg/kg rasagiline improved motor impairments, according to motor behavior testing [3].
The present study was performed to test the potential of rasagiline as a disease-modifying agent in multiple system atrophy (MSA) using a transgenic mouse model previously described by our group. (PLP)-alpha-synuclein transgenic mice featuring glial cytoplasmic inclusion pathology underwent 3-nitropropionic acid intoxication to model full-blown MSA-like neurodegeneration. Two doses of rasagiline were used (0.8 and 2.5 mg/kg) for a treatment period of 4 weeks. Rasagiline-treated animals were compared to placebo saline-treated mice by evaluation of motor behaviour and neuropathology. Motor behavioural tests including pole test, stride length test and general motor score evaluation showed improvements in motor deficits associated with 2.5 mg/kg rasagiline therapy. Immunohistochemistry and histology showed significant reduction of 3-NP-induced neuronal loss in striatum, substantia nigra pars compacta, cerebellar cortex, pontine nuclei and inferior olives of MSA mice receiving 2.5 mg/kg rasagiline. The results of the study indicate that rasagiline confers neuroprotection in a transgenic mouse model of MSA and may therefore be considered a promising disease-modifying candidate for human MSA.[3] |
| Enzyme Assay |
Determination of MAO inhibitory activity in vitro [1]
The activities of MAO-A and -B were determined by the adapted method of Tipton & Youdim (1983). Rat or human cerebral cortical tissue was homogenized in 0.3 M sucrose (one part tissue to 20 parts sucrose) using a glass-teflon motor-driven homogenizer (brain and liver), or Ultraturrax (gut). The inhibitor under test was added to a suitable dilution of the enzyme preparation in 0.05 M phosphate buffer (pH 7.4) and incubated together with selegiline 0.1 μM (for determination of MAO-A) or clorgyline 0.1 μM (for determination of MAO-B). Incubation was carried on for 60 min at 37°C before addition of labelled substrates (14C-5-hydroxytryptamine creatinine disulphate 100 μM for determination of MAO-A, or 14C-phenylethylamine 10 μM for determination of MAO-B) and incubation continued for 30 or 20 min respectively. The reaction was then stopped by addition of citric acid (2 M). Radioactive metabolites were extracted into toluene/ethyl acetate (1 : 1 v v−1), a solution of 2,5-diphenyloxazole was added to a final concentration of 0.4% (w v−1), and metabolite content estimated by liquid scintillation counting. Activity in presence of drug was expressed as a percentage of that in control samples. [1] The preincubation was carried out in the presence of clorgyline or selegiline because phenylethylamine is also metabolized quite effectively by MAO-A (O'Carroll et al., 1983), leading to inhibition curves for MAO-B, which showed a plateau at about 80% inhibition with selegiline or Rasagiline if MAO-A was not inactivated. For comparison between two inhibitors with potentially different inhibitory effects on MAO-A and MAO-B, therefore, it was thought necessary to employ the system in which opposite enzyme forms are inactivated before assay. MAO B Catalytic Activity Assay [1] SH-SY5Y and 1242-MG cells were grown to confluence, harvested, and washed with phosphate-buffered saline. One hundred micrograms of total proteins were incubated with 10 µM 14C-labeled phenylethylamine in the assay buffer (50 mM sodium phosphate buffer, pH 7.4) at 37°C for 20 min and terminated by the addition of 100 µl of 6 N HCl. The reaction products were then extracted with ethyl acetate/toluene (1:1) and centrifuged at 4°C for 10 min. The organic phase containing the reaction product was extracted, and its radioactivity was obtained by liquid scintillation spectroscopy. |
| Cell Assay |
Cell proliferation assay[2]
Cell Types: Neuroblastoma SH-SY5Y and Glioblastoma 1242-MG Tested Concentrations: 0.25 nM Incubation Duration: 96 hrs (hours) Experimental Results: Increased cell proliferation rate of SH-SY5Y cells treated with dexamethasone About 60%. The cell proliferation rate of 1242-MG cells treated with dexamethasone increased by approximately 35%. Cell Culture and Treatments [2] The SH-SY5Y and 1242-MG cells were seeded into 6-well plates and cultured overnight in medium. Cells were supplemented with charcoal-stripped, steroid-free fetal calf serum for ~6 h. The medium was then replaced with medium treated with 10 µM of dexamethasone, 0.25 nM of Rasagiline, 0.25 nM of selegiline, or 1 µM of 1-R-aminoindan in the presence of charcoal-stripped fetal calf serum. The treatments were performed every other day for 4 days. TUNEL Assay [2] The terminal deoxynucleotidyl transferase (TdT)-mediated dUTP Nick End Labeling (TUNEL) assay was used to assess the extent of apoptosis in treated cells. Briefly, cells were plated on a four-well chamber slide on the day preceding the experiment, and treated with or without 10 µM dexamethasone, 0.25 nM of Rasagiline, 0.25 nM of selegiline, or 1 µM of 1-R-aminoindan for 2 days. Cells were then washed with PBS and fixed using 4% paraformaldehyde in PBS. The slides were again washed with PBS, and fragmented DNA was detected in apoptotic cells by adding fluorescein 12-dUTP to nicked ends of DNA (In Situ Cell Death Detection Kit, Roche). Slides were incubated for 1 h at 37°C in the dark and washed in PBS three times and then visualized with a fluorescent light microscope. Green fluorescence was correlated with DNA fragmentation. Experiments were done in duplicate for three times, and the percentage of TUNEL-positive cells was determined. |
| Animal Protocol |
Animal/Disease Models: (PLP)-α-synuclein transgenic mice over 6 months old [3]
Doses: low dose (0.8 mg/kg bw) and high dose (2.5 mg/kg bw) Route of Administration: every subcutaneous injection once every 24 hrs (hrs (hours)) for 1 time. The total time was 4 weeks (from day 1 to day 28 of the experiment). Experimental Results: Low-dose treatment demonstrated no protective effect in the striatum, with neuronal numbers similar to those in placebo-treated MSA mice. High doses were associated with approximately 15% rescue of DARPP-32-immunoreactive striatal neurons. Low-dose treatment had no effect on nigral neuronal loss, but high-dose treatment completely protected nigral neurons in numbers comparable to healthy controls. Determination of inhibition of MAO activity in vivo [1] In in vivo studies, drugs were administered orally by gavage (p.o.). The animals weighed 250 – 300 g at the time of killing. For estimation of in vivo inhibitory effect, varying doses of the inhibitors were administered to groups of five or six rats for the stated times, the animals were killed by decapitation, tissues removed and frozen at −20°C, and enzyme activity determined subsequently as above. Enzyme activity in drug-treated tissues were expressed as a percentage of that in control tissues. |
| ADME/Pharmacokinetics |
Absorption, Distribution and Excretion
Rasagiline is rapidly absorbed following oral administration. The absolute bioavailability of rasagiline is about 36%. Rasagiline undergoes almost complete biotransformation in the liver prior to excretion. Glucuronide conjugation of rasagiline and its metabolites, with subsequent urinary excretion, is the major elimination pathway. After oral administration of 14C-labeled rasagiline, elimination occurred primarily via urine and secondarily via feces (62% of total dose in urine and 7% of total dose in feces over 7 days), with a total calculated recovery of 84% of the dose over a period of 38 days. Less than 1% of rasagiline was excreted as unchanged drug in urine. 87 L After oral administration of (14)C-labeled rasagiline, elimination occurred primarily via urine and secondarily via feces (62% of total dose in urine and 7% of total dose in feces over 7 days), with a total calculated recovery of 84% of the dose over a period of 38 days. Less than 1% of rasagiline was excreted as unchanged drug in urine. Rasagiline is rapidly absorbed; following oral administration, peak plasma concentrations are achieved in approximately 1 hour. The absolute bioavailability of rasagiline is about 36%. Following administration with a high-fat meal, peak plasma rasagiline concentrations and area under the plasma concentration-time curve (AUC) decreased by approximately 60 and 20%, respectively; because AUC is not substantially affected, rasagiline may be administered with or without food. Rasagiline readily crosses the blood-brain barrier. The mean steady-state or terminal half-life of rasagiline is 31 or 1.342 hours, respectively; however, there is no correlation between rasagiline's pharmacokinetic profile and its pharmacologic effects because the drug irreversibly inhibits MAO-B, and restoration of normal enzyme activity depends on the rate of de novo enzyme synthesis. Rasagiline is approximately 88-94% bound to plasma proteins, with 61-63% bound to albumin. IV studies in rats and dogs show that the volume of distribution (Vd) of rasagiline is several times that of total body water, indicating extensive tissue distribution. Tissue distribution of (14)C-rasagiline was studied in albino and pigmented rats, revealing peaks of tissue radioactivity between 0.25 and 0.5 hours. Distribution to large intestine, urinary bladder and lacrimal glands takes longer, whilst persistence (up to 24 hrs) was seen in eyes, skin and arterial walls of pigmented animals. In-vitro protein binding in plasma of animals is in the range of 70 to 90% and in human plasma in the range of 88 to 94%. Oral studies with (14)C-rasagiline show that absorption is rapid in all species, with Cmax attained in less than 2 hours. Absolute bioavailability has been estimated as 53-69% in rats, 13-22% in dogs, and 36% in humans. Toxicokinetic analyses during the toxicology studies showed that exposure was linear at doses higher than the pharmacological selectivity for inhibition of MOA-B and was maintained up to about 5 mg/kg/day. However, kinetics became non-linear at higher doses, possibly indicating saturation of the elimination processes for both rasagiline and its metabolite aminoindan. Accumulation was seen only at the highest doses in the mouse and dog studies (60 and 21 mg/kg/day respectively). For more Absorption, Distribution and Excretion (Complete) data for RASAGILINE (6 total), please visit the HSDB record page. Metabolism / Metabolites Rasagiline undergoes almost complete biotransformation in the liver prior to excretion. In vitro experiments indicate that both routes of rasagiline metabolism are dependent on the cytochrome P450 (CYP) system, with CYP 1A2 being the major isoenzyme involved in rasagiline metabolism. Rasagiline is extensively metabolized in the liver following oral administration. In vitro studies have shown that CYP1A2 is the predominant P450 isoform involved in the metabolic elimination of rasagiline. The primary human plasma metabolite formed following biotransformation of rasagiline is aminoindan. The proposed principal biotransformation pathways of rasagiline in human are N-dealkylation, hydroxylation of the indan ring, along with Phase II N or O-conjugation, including N-glucuronidation of the parent drug and of its metabolites. There was no bioconversion of rasagiline mesylate (R enantiomer) to its S enantiomer within the human body, as determined in plasma samples for healthy volunteers dosed with rasagiline. Rasagiline is not metabolized to amphetamine or methamphetamine. An extensive first pass metabolism effect is evident, likely due to rasagiline binding to MAO sites in the intestine prior to passing the liver. Metabolism is rapid and extensive, with a similar profile in all tested species. The primary route of biotransformation is via N-dealkylation to form aminoindan and by hydroxylation to form 3-hydroxy-N-propargyl-1-aminoindan. Conjugation by sulfide or glucuronic acid occurs. Microsomal studies indicate CYP1A2 as the primary metabolising isotype, but rasagiline is neither an inducer nor inhibitor of cytochrome p450. The metabolism of rasagiline under inhibition, induction of CYP1A2 or in presence of concomitant substrate to the enzyme has been addressed clinically. Rasagiline undergoes almost complete biotransformation in the liver prior to excretion. The metabolism of rasagiline proceeds through two main pathways: N-dealkylation and/or hydroxylation to yield 1-aminoindan (AI), 3-hydroxy-N-propargyl-1 aminoindan (3-OH-PAI) and 3-hydroxy-1-aminoindan (3-OH-AI). In vitro experiments indicate that both routes of rasagiline metabolism are dependent on the cytochrome P450 (CYP) system, with CYP 1A2 being the major isoenzyme involved in rasagiline metabolism. Glucuronide conjugation of rasagiline and its metabolites, with subsequent urinary excretion, is the major elimination pathway. Biological Half-Life Rasagiline has a mean steady-state half life of 3 hours but there is no correlation of pharmacokinetics with its pharmacological effect because of its irreversible inhibition of MAO-B. Rasagiline's mean steady-state half life is 3 hours ... . Rasagiline is eliminated with a half life of about 0.6 - 2 hours and ranging from 0.3 to 3.5 hours across the 0.5 to 20 mg dose range examined following oral administration. |
| Toxicity/Toxicokinetics |
Hepatotoxicity
Rasagiline has been reported to cause serum enzyme elevations in a small proportion of patients treated long term, although the abnormalities were usually mild and self-limiting. Rasagiline has not been implicated in cases of acute liver injury, but such instances have been reported with other less specific MAO inhibitors. Likelihood score: E (unlikely cause of clinically apparent liver injury). Effects During Pregnancy and Lactation ◉ Summary of Use during Lactation No clinical use of rasagiline during breastfeeding has been reported. Rasagiline might reduce serum prolactin and interfere with milk production. An alternate drug may be preferred, especially while nursing a newborn or preterm infant. ◉ Effects in Breastfed Infants Relevant published information was not found as of the revision date. ◉ Effects on Lactation and Breastmilk Animal studies show that rasagiline reduces serum prolactin. The clinical relevance of these findings in nursing mothers is not known. The prolactin level in a mother with established lactation may not affect her ability to breastfeed. Protein Binding Plasma protein binding ranges from 88-94% with mean extent of binding of 61-63% to human albumin over the concentration range of 1-100 ng/ml. Interactions Antidepressant Agents, Selective Serotonin-reuptake Inhibitors (SSRIs): Potential pharmacologic interaction resembling serotonin syndrome (hyperthermia, rigidity, myoclonus, autonomic instability with rapid vital sign fluctuations, and mental status changes that may progress to extreme agitation, delirium, coma, and death). Concomitant use generally should be avoided. At least 14 days should elapse between discontinuance of rasagiline and initiation of an SSRI.Because both fluoxetine and its principal metabolite have relatively long half-lives, the manufacturer of rasagiline recommends that at least 5 weeks (or longer with high-dose or long-term fluoxetine therapy) elapse between discontinuance of fluoxetine therapy and initiation of rasagiline. Inhibitors of CYP1A2: Pharmacokinetic interaction observed during concomitant use with ciprofloxacin (increased plasma rasagiline concentrations). Dosage of rasagiline should be limited ... in patients receiving the drug concomitantly with ciprofloxacin or other CYP1A2 inhibitors. St. John's wort (Hypericum perforatum): Concomitant use is contraindicated. Potential pharmacologic interaction (resembling serotonin syndrome) with meperidine (coma, severe hypertension or hypotension, severe respiratory depression, seizures, malignant hyperpyrexia, excitation, peripheral vascular collapse, and death). Concomitant use with meperidine, methadone, propoxyphene, or tramadol is contraindicated. At least 14 days should elapse between discontinuance of rasagiline and initiation of meperidine. For more Interactions (Complete) data for RASAGILINE (12 total), please visit the HSDB record page. |
| References |
|
| Additional Infomation |
Therapeutic Uses
Rasagiline is used as initial monotherapy or as adjunctive therapy to levodopa for the symptomatic treatment of idiopathic parkinsonian syndrome. Azilect (rasagiline mesylate) is indicated for the treatment of the signs and symptoms of idiopathic Parkinson's disease as initial monotherapy and as adjunct therapy to levodopa. The effectiveness of Azilect was demonstrated in patients with early Parkinson's disease who were receiving Azilect as monotherapy and who were not receiving any concomitant dopaminergic therapy. The effectiveness of Azilect as adjunct therapy was demonstrated in patients with Parkinson's disease who were treated with levodopa. Drug Warnings When used as monotherapy, postural hypotension was reported in approximately 3% of patients treated with 1 mg rasagiline and 5% of patients treated with placebo. In the monotherapy trial, postural hypotension did not lead to drug discontinuation and premature withdrawal in the rasagiline or placebo treated patients. When used as an adjunct to levodopa, postural hypotension was reported in approximately 6% of patients treated with 0.5 mg rasagiline, 9% of patients treated with 1 mg rasagiline and 3% of patients treated with placebo. Postural hypotension led to drug discontinuation and premature withdrawal from clinical trials in one (0.7%) patient treated with rasagiline 1 mg/day, no patients treated with rasagiline 0.5 mg/day and no placebo-treated patients. Clinical trial data suggest that postural hypotension occurs most frequently in the first two months of rasagiline treatment and tends to decrease over time. Data from epidemiologic studies indicate that patients with Parkinson's disease have an approximately twofold to fourfold greater risk of developing melanoma than the general population; however, it is unclear whether the observed increased risk is related to the underlying disease or to antiparkinsonian drug therapy. The risk of developing melanoma in patients receiving rasagiline appears to be greater than that in the general population but comparable to that in patients with Parkinson's disease. Because of these findings, patients and clinicians should monitor for melanomas frequently. The manufacturer recommends that dermatologic examinations be performed by qualified clinicians (e.g., dermatologists) periodically; the frequency of dermatologic examinations should be determined by the patient's dermatologist. In the monotherapy study, hallucinations were reported as an adverse event in 1.3% of patients treated with 1 mg rasagiline and in 0.7% of patients treated with placebo. In the monotherapy trial, hallucinations led to drug discontinuation and premature withdrawal from clinical trials in 1.3% of the 1 mg rasagiline treated patients and in none of the placebo treated patients. When used as an adjunct to levodopa, hallucinations were reported as an adverse event in approximately 5% of patients treated with 0.5 mg/day, 4% of patients treated with 1 mg/day rasagiline and 3% of patients treated with placebo. Hallucinations led to drug discontinuation and premature withdrawal from clinical trials in about 1% of patients treated with 0.5 mg/day or 1 mg/day and none of the placebo treated patients. Patients should be cautioned of the possibility of developing hallucinations and instructed to report them to their health care provider promptly should they develop. Safety and efficacy of rasagiline have not been established in pediatric patients younger than 18 years of age. For more Drug Warnings (Complete) data for RASAGILINE (10 total), please visit the HSDB record page. Pharmacodynamics Rasagiline is a propargylamine and an irreversible inhibitor of monoamine oxidase (MAO). MAO, a flavin-containing enzyme, regulates the metabolic degradation of catecholamines and serotonin in the CNS and peripheral tissues. It is classified into two major molecular species, A and B, and is localized in mitochondrial membranes throughout the body in nerve terminals, brain, liver and intestinal mucosa. MAO-A is found predominantly in the GI tract and liver, and regulates the metabolic degradation of circulating catecholamines and dietary amines. MAO-B is the major form in the human brain and is responsible for the regulation of the metabolic degradation of dopamine and phenylethylamine. In ex vivo animal studies in brain, liver and intestinal tissues rasagiline was shown to be a potent,selective, and irreversible monoamine oxidase type B (MAO-B) inhibitor. At the recommended therapeutic doses, Rasagiline was also shown to be a potent and irreversible inhibitor of MAO-B in platelets. The selectivity of rasagiline for inhibiting only MAO-B (and not MAO-A) in humans and the sensitivity to tyramine during rasagiline treatment at any dose has not been sufficiently characterized to avoid restriction of dietary tyramine and amines contained in medications. Drug Indication Azilect is indicated for the treatment of idiopathic Parkinson's disease (PD) as monotherapy (without levodopa) or as adjunct therapy (with levodopa) in patients with end-of-dose fluctuations. The present study has demonstrated that rasagiline, like selegiline, is an irreversible inhibitor of MAO-B. This was demonstrated in experiments where rasagiline MAO inhibitory activity was examined in vitro and in vivo when it was given p.o. and MAO-A and -B were then estimated ex vivo in various tissues, at time intervals up to 13 days after rasagiline treatment. It is apparent that rasagiline is a very potent selective MAO-B inhibitor and has a good uptake across the blood-brain barrier, as shown by the similarity of inhibition curves between liver and brain. Although when compared in vitro, rasagiline had similar potency to selegiline for inhibition of MAO-B, the in vivo study showed a greater potency of rasagiline. This greater potency of rasagiline is even more marked if, instead of 50% enzyme inhibition, the dose required for 80% inhibition is measured. The reason for this is not currently known, but may be due to different rates of metabolism of the parent compounds in vivo, or to improved tissue penetration of rasagiline. Interestingly, preliminary studies in humans show an approximately 5 fold greater potency for rasagiline over selegiline for inhibition of platelet MAO-B (unpublished data). Although rasagiline has a greater potency than selegiline, its selectivity for MAO-A and -B inhibition is very similar to what has been reported for selegiline. However, in contrast to selegiline which does not show selectivity between its optical isomers for inhibition of MAO-A and -B, AGN 1135 shows roughly 4 and 2 orders of magnitude between its optical isomers for inhibition of MAO-B and -A respectively. The present results complement findings in non-human primate (monkey) brains (Gotz et al., 1998) where rasagiline was given chronically for 7 days at various doses and MAO-A and -B activities were measured in several brain regions, including caudate nucleus, globus pallidus, cerebral cortex and hippocampus. Rasagiline was shown to be a potent selective inhibitor of MAO-B in the caudate nucleus and globus pallidus where the activity of MAO-B is 4 fold higher than that of MAO-A (Gotz et al., 1998). The recovery of the MAO-A and -B activities after in vivo inhibition, which is related to the synthesis of enzyme apoprotein, differs between the tissues (liver, intestine and brain) examined. The small intestine MAO-B activity has the fastest recovery, while the brain MAO-B activity shows the slowest recovery. These differences in rat tissue enzyme activity recovery after rasagiline treatment, are not unusual since similar findings have been reported for enzyme recovery after inhibition by selegiline and clorgyline (Neff & Goridis, 1972; Della Corte & Tipton, 1980). Indeed, in primate (monkeys and human) brains the half-life for recovery of MAO-B after selegiline treatment has been reported to be well over 30 days (Fowler et al., 1994), and for the rat brain 13 days (Neff & Goridis, 1972; Della Corte & Tipton, 1980). In conclusion, the present study has shown that rasagiline is a potent irreversible inhibitor of MAO-B and it is 3 – 15 times more potent than selegiline in the rat in vivo with a similar selectivity for inhibition of MAO-B to MAO-A. Because of its cleaner pharacological profile, with absence of amphetamine-like properties, formation of the metabolite aminoindan rather than 1-methamphetamine, and recently described neuroprotective and antiapoptotic properties (Finberg et al., 1998; Huang et al., 1999; Youdim et al., 1999), we can conclude that this drug may have a preferential activity to that of selegiline in the treatment of Parkinson's disease.[1] We report here for the first time that rasagiline, selegiline, and 1-R-aminoindan significantly prevent dexamethasone-induced brain cell death involving in both neuroblastoma and glioblastoma cells. Among the three compounds, rasagiline has the highest neuroprotective effect compared to either selegiline or 1-R-aminoindan. Rasagiline (Azilect) and selegiline (1-deprenyl or Emsam) are irreversible inhibitors of MAO B. The greater neuroprotective quality of rasagiline may in part be due to the effects of the parent compound and its major metabolite, 1-R-aminoindan. Furthermore, the inhibitory effects of these drugs on MAO B catalytic activity and on apoptotic DNA fragment damage (observed by TUNEL staining) were examined. Rasagiline has shown the highest inhibition on MAO B enzymatic activity (Youdim et al. 2001a) and also has shown the highest prevention on apoptosis compared to selegiline and 1-R-aminoindan. The mechanism by which rasagiline and selegiline initiate their anti-apoptotic effect can be summarized by their up regulation of anti-apoptotic Bcl-2 and Bcl-Xl and down regulation of propaoptotic Bad, Bax, PARP, and caspase 3 (see Youdim et al. 2005a) and Youdim et al. 2006) for reviews). Because Bcl-2 and caspase 3 are key factors for preventing or mediating the mitochondrial-involved apoptosis (Lakhani et al. 2006), it suggests that the MAO inhibitors may protect cells from apoptosis through a mechanism involving the maintenance of mitochondrial homeostasis (Malorni et al. 1998). In addition, structure activity studies with rasagiline have shown that it is propargylamine moiety that produces this effect, since propargylamine which has little or no MAO inhibitory activity has a similar mechanism of neuroprotective activity with similar potency (Bar-Am et al. 2005). Furthermore, both rasagiline and proppargylamine activate neuroprotective protein kinase C (PKCα and PKCε), while down-regulating propaoptotic PKCδ and γ. Inhibition of PKC by GF109203X prevents their neuroprotective activity (Weinreb et al. 2005; Youdim et al. 2005a). The mechanism by which aminoindan has been fully elucidated. The neuroprotective properties of 1-R-aminoindan have been assessed employing a cytotoxic model of human neuroblastoma SKN-SH cells in high-density culture-induced neuronal death and in response to 6-hydroxydopamine. We show that 1-R-aminoindan (0.1–1 µM) significantly reduced the apoptosis-associated phosphorylated protein, H2A.X (Ser139), decreased the cleavage of caspase 9 and caspase 3, while increasing the anti-apoptotic proteins, Bcl-2 and Bcl-xl. Protein kinase C (PKC) inhibitor, GF109203X, prevented the neuroprotection, indicating the involvement of PKC in aminoindan-induced cell survival. Aminoindan markedly elevated pPKC (pan) and specifically that of the pro-survival PKC isoform, PKC epsilon (Bar-Am et al. 2007). In summary, the neurorptoective activity seen with rasagiline and its major metabolite, 1-R-aminoindan in the present and previous studies, may have relevance to the recent prospective clinical study in Parkinsonian subjects, ADAGIO, where rasagiline indicated that early treatment with rasagiline provided benefits that were not obtained with later initiation of the drug. This is the first time that a prospective large-scale, randomized, double-blind trial has provided evidence that a drug might slow down PD progression via neuroprotection (Hughes 2008).[2] |
| Molecular Formula |
C12H13N
|
|---|---|
| Molecular Weight |
171.2383
|
| Exact Mass |
171.104
|
| Elemental Analysis |
C, 84.17; H, 7.65; N, 8.18
|
| CAS # |
136236-51-6
|
| Related CAS # |
Rasagiline mesylate;161735-79-1;Rasagiline-13C3 mesylate racemic;1216757-55-9
|
| PubChem CID |
3052776
|
| Appearance |
Light yellow to brown <38°C powder,>41°C liquid
|
| Density |
1.1±0.1 g/cm3
|
| Boiling Point |
305.5±30.0 °C at 760 mmHg
|
| Melting Point |
35-41°C
|
| Flash Point |
146.8±20.0 °C
|
| Vapour Pressure |
0.0±0.6 mmHg at 25°C
|
| Index of Refraction |
1.577
|
| LogP |
2.27
|
| Hydrogen Bond Donor Count |
1
|
| Hydrogen Bond Acceptor Count |
1
|
| Rotatable Bond Count |
2
|
| Heavy Atom Count |
13
|
| Complexity |
212
|
| Defined Atom Stereocenter Count |
1
|
| SMILES |
N([H])(C([H])([H])C#C[H])[C@@]1([H])C2=C([H])C([H])=C([H])C([H])=C2C([H])([H])C1([H])[H]
|
| InChi Key |
RUOKEQAAGRXIBM-GFCCVEGCSA-N
|
| InChi Code |
InChI=1S/C12H13N/c1-2-9-13-12-8-7-10-5-3-4-6-11(10)12/h1,3-6,12-13H,7-9H2/t12-/m1/s1
|
| Chemical Name |
(1R)-N-prop-2-ynyl-2,3-dihydro-1H-inden-1-amine
|
| Synonyms |
rasagiline; 136236-51-6; (R)-N-(2-Propynyl)-2,3-dihydroinden-1-amine; Azilect; (R)-N-2-Propynyl-1-indanamine; (1R)-N-(prop-2-yn-1-yl)-2,3-dihydro-1H-inden-1-amine; 1-Indanamine, N-2-propynyl-, (R)-; TV-1030;
|
| HS Tariff Code |
2934.99.9001
|
| Storage |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| Solubility (In Vitro) |
DMSO : ~100 mg/mL (~583.98 mM)
H2O : ≥ 5.88 mg/mL (~34.34 mM) |
|---|---|
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.5 mg/mL (14.60 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution.
For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2.5 mg/mL (14.60 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. View More
Solubility in Formulation 3: ≥ 2.5 mg/mL (14.60 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 5.8398 mL | 29.1988 mL | 58.3976 mL | |
| 5 mM | 1.1680 mL | 5.8398 mL | 11.6795 mL | |
| 10 mM | 0.5840 mL | 2.9199 mL | 5.8398 mL |
*Note: Please select an appropriate solvent for the preparation of stock solution based on your experiment needs. For most products, DMSO can be used for preparing stock solutions (e.g. 5 mM, 10 mM, or 20 mM concentration); some products with high aqueous solubility may be dissolved in water directly. Solubility information is available at the above Solubility Data section. Once the stock solution is prepared, aliquot it to routine usage volumes and store at -20°C or -80°C. Avoid repeated freeze and thaw cycles.
Calculation results
Working concentration: mg/mL;
Method for preparing DMSO stock solution: mg drug pre-dissolved in μL DMSO (stock solution concentration mg/mL). Please contact us first if the concentration exceeds the DMSO solubility of the batch of drug.
Method for preparing in vivo formulation::Take μL DMSO stock solution, next add μL PEG300, mix and clarify, next addμL Tween 80, mix and clarify, next add μL ddH2O,mix and clarify.
(1) Please be sure that the solution is clear before the addition of next solvent. Dissolution methods like vortex, ultrasound or warming and heat may be used to aid dissolving.
(2) Be sure to add the solvent(s) in order.
| NCT Number | Recruitment | interventions | Conditions | Sponsor/Collaborators | Start Date | Phases |
| NCT03727139 | Completed Has Results | Drug: Rasagiline | Parkinson's Disease | Takeda | November 1, 2018 | |
| NCT01879748 | Completed | Drug: Rasagiline Drug: Placebo |
Parkinson's Disease | Teva Branded Pharmaceutical Products R&D, Inc. |
June 2013 | Phase 1 |
| NCT01032486 | Completed | Drug: Rasagiline mesylate | Parkinson's Disease | Teva Branded Pharmaceutical Products R&D, Inc. |
December 2009 | |
| NCT00203164 | Completed | Drug: rasagiline mesylate | Parkinson's Disease | Teva Branded Pharmaceutical Products R&D, Inc. |
May 2002 | Phase 3 |
|
|
|
Products are for research use only; We do not sell to patients
Copyright 2020 InvivoChem LLC | All Rights Reserved